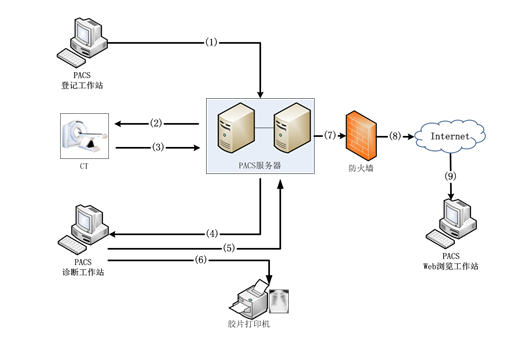
|
标号 |
说明 |
|
1 |
医生使用PACS登记工作站录入患者信息,包括:姓名、图像号、性别、设备、部位等。并将这些信息提交到PACS服务器中进行存储。 |
|
2 |
CT等设备的采集工作站,通过DICOM Modality Worklist协议,从PACS服务器获取已经登记的、等待检查的患者信息。例如:患者姓名、图像号、设备、部位等。 |
|
3 |
CT等设备对患者进行图像采集时,通过DICOM MPPS协议向PACS服务器发送当前的检查状态。
CT等设备生成图像后,通过DICOM Storage协议将带有患者信息的DICOM格式图像发送给PACS服务器。
PACS服务器接收到图像后,将图像存到服务器的磁盘阵列中,将检查信息补充到数据库中,并通过DICOM Storage Commitment协议告知CT等设备图像的存储状态。 |
|
4 |
医生使用PACS诊断工作站进行图像浏览,通过DICOM Query/Retrieve协议从PACS服务器上将患者信息、图像数据下载到本机磁盘,进行诊断并编写报告。 |
|
5 |
医生在PACS诊断工作站编写完报告后,将报告内容提交给PACS服务器进行存储。 |
|
6 |
医生在PACS诊断工作站对胶片进行排版后,通过DICOM Print协议,将胶片发给胶片打印机进行打印。 |
|
7~9 |
医生使用本系统的PACS Web浏览工作站对患者信息、图像数据进行查看时,PACS服务器通过DICOM WADO协议,将这些数据通过互联网传输到用户的浏览器中。 |