|
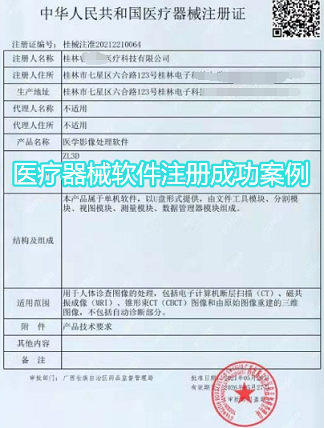
医疗器械独立软件注册如何制定性能指标,检验报告需要包括哪些内容?
答:独立软件可以直接参考《医疗器械软件注册技术审查指导原则》附录I的模板进行编写产品技术要求。对于医学图像存储传输软件(PACS),还需明确图像传输后图像数据的一致性和完整性,明确在指定测试条件下打开图像的时间(包含图像传输与显示的时间),测试条件应明确网络环境是单机、局域网或广域网。
预期用于诊断的移动图像处理软件,根据《移动医疗器械注册技术审查指导原则》,产品技术要求制定具有数据不可得性的功能、产品注册信息用户确认功能、运行环境开机自检功能、环境光检测功能、显示屏亮度矫正功能等性能指标。
独立软件检验报告还需要包含软件完整版本和软件发布版本的界面照片。如有多个运行环境或多个发布版本,则每个互不兼容的运行环境或每个互不涵盖的发布版本均应作为一个检测单元。比如,操作系统软件包含Windows和iOS,检验报告需分别检测。对于Web浏览器,所支持的互不兼容的浏览器(如IE、Chrome、Firefox等)应分别作为一个检测单元。
医疗器械软件注册描述文档如何进行编写,软件研究资料需要注意哪些问题?
答:软件描述文档根据《医疗器械软件注册技术审查指导原则》“表1 软件描述文档框架”的要求进行编写。比如,软件安全性级别为B级的产品,除了软件描述文档正文外,一般还包括以下附件:软件需求规范全文;软件开发生存周期计划、配置管理计划和维护计划的摘要;系统测试、用户测试的计划与报告。
此外,还需出具软件版本命名规则真实性声明,明确软件版本的全部字段及字段含义,确认软件完整版本和软件发布版本。
医疗器械注册申请人声称产品符合DICOM标准,则应提供DICOM符合性声明,DICOM符合性声明的编写方法和内容参照DICOM标准中的相关规定。
若医疗器械产品注册申请人声明产品符合HL7标准,则应提供HL7符合性声明,HL7符合性声明的编写方法和内容参照HL7标准中的相关规定。
深圳鸿远医疗器械咨询有限公司是一家技术专业的医疗器械咨询公司,专注提供全国各地如:深圳、广州、东莞、中山、佛山、潮州、顺德、广西、上海、西安、重庆、成都等知名城市的医疗器械领域技术咨询服务。鸿远医疗器械咨询专业服务于:医疗器械注册证、医疗器械生产许可证、进口医疗器械注册、一类医疗器械产品备案及生产备案代办、医疗器械经营许可证代办、二类医疗器械经营备案、医疗器械分类界定、CE认证、ISO13485认证、FDA注册、临床试验、医疗器械质量管理体系认证及体系建立与过程确认文件建立(ISO9001, ISO13485, GMP, CE,QSR820,CMDCAS);产品技术要求制订、技术文件、临床试验及免临床同类产品比对比资料编写、注册资料编写辅导、电磁兼容预测整改、医疗器械广告批文申报、医疗器械出口销售证明办理、洁净室建设指导等提供一站式服务,技术专业,诚信服务,欢迎您咨询!
|

