|
有源植入医疗器械CE认证咨询
CE为法文CONFORMITE EUROPEENNE的字首缩写,表示“欧洲统一”。“CE”标志是一种安全认证标志,凡是贴有“CE”标志的产品就可在欧盟各成员国内销售,无须符合每个成员国的要求,被视为制造商打开并进入欧洲市场的护照。使用CE标志,实现了商品在欧盟成员国范围内的自由流通。
在欧盟市场“CE”标志属强制性认证标志,不论是欧盟内部企业生产的产品,还是其他国家生产的产品,要想在欧盟市场上自由流通,就必须加贴“CE”标志,以表明产品符合欧盟《技术协调与标准化新方法》指令的基本要求。这是欧盟法律对产品提出的一种强制性要求。
有源植入性医疗器械的CE认证按欧盟指令90/385/EEC的要求进行。该指令于1993年1月1日生效,1995年1月1日强制执行。
所谓有源植入性医疗器械,是指任何通过外科或内科手段,拟部分或全部插入人体,或通过医疗手段介入自然腔口且拟留在体内的有源医疗器械。而有源医疗器械是指任何依靠电能或其它能源而不是直接由人体或重力产生的能源来发挥其功能的医疗器械。
有源植入医疗器械指令不适用于下列产品:
(a) 第2001/83//EEC号指令覆盖的药品;
(b) 人血、人血制品、人血浆或人血细胞,以及与上述制品组合投放市场的器械;
(c) 人体移植物,组织或细胞以及人体组织或细胞的组合或衍生制品;
(d) 动物体移植物,组织或细胞,但利用死的动物组织或从动物组织中衍生的不能存活的产品制造的器械不在此列。
有源植入式医疗器械进行CE认证时的符合性评估,主要包括两条主要的途径:
1)全面质量管理体系评估途径;
2)EC型式试验途径,包括EC型式试验+生产质量保证体系;及EC型式试验+EC验证两种分支途径;
有源植入医疗器械CE认证的重点在于:
型式检验
产品设计检验
全面质量体系评估
医疗器械临床试验
其他专项要求(电气安全,电磁兼容, 生物兼容性,产品寿命等)
有源植入医疗器械CE认证的一般步骤:
步骤1. 分析该器械的特点,确定其属于有源医疗器械指令范围
步骤2. 选择相应的符合性评价程序
步骤3. 选择一个公告机构
步骤4. 确认适用的基本要求/有关的协调标准
步骤5. 确认该器械满足基本要求/协调标准, 并使证据文件化
步骤6. 对于需要公告机构评审的器械,通过公告机构的符合性程序
步骤7. 起草符合性声明并加贴CE标志
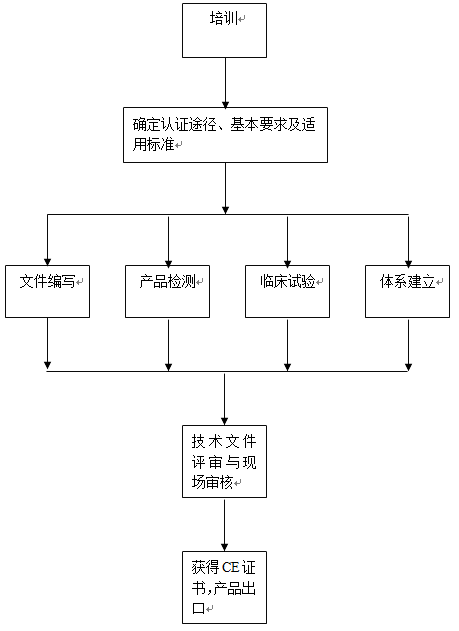
有源植入医疗器械CE认证咨询服务流程:
|