|
什么是医疗器械FDA注册认证? 2017年医疗器械FDA注册认证多少钱? 医疗器械FDA认证周期多长?
FDA对医疗器械有明确和严格的定义,其定义如下:“所谓医疗器械是指符合以下条件之仪器、装置、工具、机械、器具、插入管、体外试剂及其它相关物品,包括组件、零件或附件:明确列于National Formulary或the Unite States Pharmacopeia或前述两者的附录中者;预期使用于动物或人类疾病,或其它身体状况之诊断,或用于疾病之治愈、减缓与治疗者;预期影响动物或人体身体功能或结构,但不经由新陈代谢来达到其主要目的者”。
只有符合以上定义的产品方被看作医疗器械,在此定义下,不仅医院内各种仪器与工具,即使连消费者可在一般商店购买之眼镜框、眼镜片、牙刷与按摩器等健身器材等都属于FDA之管理范围。它与国内对医疗器械的认定稍有不同。
根据风险等级的不同,FDA注册将医疗器械分为三类(Ⅰ,Ⅱ,Ⅲ),Ⅲ类风险等级最高。FDA将每一种医疗器械都明确规定其产品分类和管理要求,FDA医疗器械产品目录中共有1,700多种。任何一种医疗器械想要进入美国市场,必须首先弄清申请上市产品分类和管理要求。
FDA针对医疗器械制订了许多法案,并不时地进行修改和补充,但根本的法案并不多,主要包括:联邦食品、药品与化妆品法案(FD&C Act,根本法案);公众健康服务法案;公正包装和标识法案;健康和安全辐射控制法案;安全医疗器械法案;现代化法案。对这些法案,FDA认证给予了非常详细的解释,并配套有具体的操作要求。企业在计划进入美国市场前,需仔细评估针对自己产品相关的法规和具体要求(包括不同的美国产品标准要求)。
在明确了以上信息后,企业就可以着手准备有关的申报资料,并按一定程序向FDA申报以获取批准认可。对于任何产品,企业都需进行企业注册(Registration)和产品列名(Listing)。对Ⅰ类产品(占47%左右),实行的是一般控制(General Control),绝大部分产品只需进行注册、列名和实施GMP规范,产品即可进入美国市场(其中极少数产品连GMP也豁免,极少数保留产品则需向FDA递交510(K)申请即PMN(Premarket Notification));对Ⅱ类产品(占46%左右),实行的是特殊控制(Special Control),企业在进行注册和列名后,还需实施GMP和递交510(K)申请(极少产品是510(K)豁免);对Ⅲ类产品(占7%左右),实施的是上市前许可,企业在进行注册和列名后,须实施GMP并向FDA递交PMA(Premarket Application)申请(部分Ⅲ类产品还是PMN)。
对Ⅰ类产品,企业向FDA认证递交相关资料后,FDA只进行公告,并无相关证件发给企业;对Ⅱ、Ⅲ类器械,企业须递交PMN或PMA,FDA在公告的同时,会给企业以正式的市场准入批准函件(Clearance),即允许企业以自己的名义在美国医疗器械市场上直接销售其产品。至于申请过程中是否到企业进行现场GMP考核,则由FDA根据产品风险等级、管理要求和市场反馈等综合因素决定。
2017年医疗器械FDA注册认证收费标准:
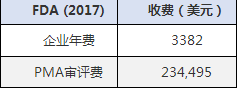
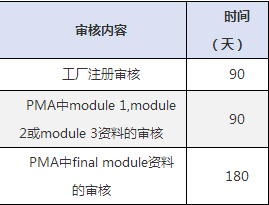
医疗器械FDA注册认证周期:
深圳鸿远医疗器械咨询有限公司 http://www.hongyuanyixiezixun.com专业代办医疗器械产品注册证咨询代理、医疗器械生产许可证、三类医疗器械经营许可证、二类医疗器械经营备案、进口医疗器械注册、一类医疗器械产品备案及生产备案、FDA注册、ISO13485认证、 CE认证、计量器具生产许可证、临床试验、出口证,自由销售证等代理代办、医疗器械质量管理体系认证文件的建立及体系与过程确认文件的建立 (如:ISO9001、 ISO13485 、GMP、 CE、QSR820、CMDCAS)产品检测,临床试验及免临床资料编写、产品技术要求制订、技术文件编写辅导、医疗器械广告批文申请办理、洁净室建设指导等服务,技术专业,诚信服务,代理费用低,欢迎您咨询!
|

