|
医疗器械牙科纤维桩产品临床评价资料要求申请人应按《医疗器械注册管理办法》、《医疗器械临床评价技术指导原则》选择合理的临床评价方式,提交临床评价资料。
1.依据《免于进行临床试验的第三类医疗器械目录》,对于符合YY/T 0517的相关要求,用于根管治疗后牙体大面积缺损时对重建的冠核进行可靠固位的牙科纤维桩产品,免于进行临床试验。豁免情况不包括使用了新材料、活性成分、新技术、新设计或具有新作用机理、新功能的产品。
2.临床试验基本要求
在中国境内开展临床试验的应符合《医疗器械临床试验质量管理规范》及本指导原则的要求。应对产品是否满足使用要求或者适用范围进行确认。提交的临床评价资料应当包括临床试验协议、临床试验方案和临床试验报告等。
根据牙科纤维桩产品使用特点,纤维桩的有效性需通过桩核冠(纤维桩-树脂核-冠/桥)修复体进行整体验证,冠桥修复体需经由临床机构设计并经技工室加工制作,即纤维桩的有效性不仅取决于产品本身属性,还与配套使用粘接剂、树脂核材料属性及桩核冠修复体的临床设计、加工工艺、临床操作水平有关,因此建议牙科纤维桩产品的临床试验应满足合理的修复体设计和规范的临床操作的要求。
临床试验时应注意如下几方面,供参考:
(1)受试对象
根据本产品目标适用人群,在临床研究方案中描述受试者口腔状况并规定完善合理的受试者纳入标准及排除标准。
受试者口腔状况至少应描述:口腔卫生状况,牙齿清洁度及色泽,牙龈及口腔黏膜情况,根尖周组织和牙周组织情况,咬合情况,牙体缺损情况,牙列缺损及缺失情况,预修复的牙位,根管充填情况,修复体的类型,使用辅助材料的情况等。
参考纳入标准为:牙冠有缺损,需使用桩核冠修复;患牙已行完善根管治疗,无根尖病变;牙槽骨无明显吸收,牙根长度大于等于临床冠长度;有完整牙本质肩领,不松动,无叩痛;口腔卫生良好,牙龈健康无红肿出血;行单冠修复,不作为固定桥或活动义齿的基牙。对不符合纳入标准的入选者应予剔除。
(2)对照组选择及试验设计类型
牙科纤维桩产品的临床试验根据需要可设置已上市产品作为对照组,建议尽可能采用随机对照试验,并明确比较类型(优效检验、等效或非劣效检验等)。如属于非劣效性检验、等效性检验或优效性检验的比较类型,应事先规定具有临床意义的界值。如采用单组目标值法,应事先明确目标值及其确定依据。
(3)评价指标及评价标准
①评价指标
为评价被试产品的性能,应明确临床试验的主要疗效评价指标、次要疗效评价指标及安全性评价指标。
有效性评价指标设定举例:桩核边缘完整性、桩核松动、桩核折断、桩核脱落、全冠脱落、牙根劈裂、继发龋、根尖病变、患牙叩痛、牙周情况等。对于新型牙科纤维桩,还应根据其宣称的适应症和性能等增加相应的评价项目。
有效性评价指标设定举例:不良反应、不良事件及并发症、牙科检查、生命体征等。
②评价标准
评价指标需采用国际公认的评价标准,如果无公认的标准,需采用临床常规疗效评价标准。
牙科纤维桩产品临床评价标准示例如下(表1)(供参考):
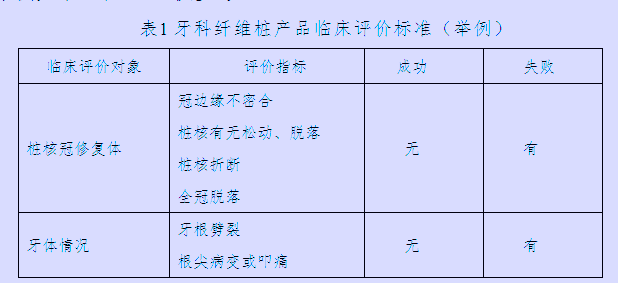
医疗器械临床试验过程中,还应记录各组患者的就诊次数和操作时间,记录在临床研究期间任何可能影响试验结果的药物治疗及服用/使用剂量,如抗生素、镇痛剂、漱口水等的用药情况等。
(4)临床观察
①修复前评估
修复前评估应包含:受试者的全身状况,可能影响试验结果的任何疾病情况,受试者口腔状况,包括:牙齿缺损程度、根面位置、牙根长度、直径和形状、根管充填情况、根尖病变愈合情况、牙周膜有无增宽情况、牙周健康状况,牙槽骨吸收程度等。
②临床操作步骤
应详细记录临床的操作步骤,明确所选用纤维桩、核树脂、纤维桩桩道预备工具及粘接剂系统的产品名称型号等。至少应记录以下操作步骤和要点:
冠部初步预备:去除修复牙齿腐质或原充填体的情况,牙本质肩领高度及厚度等。
桩道预备:X线片确定的纤维桩在根管内长度,配套用引导钻、扩孔钻、终钻型号,预备过程,预备完成后X线片显示的桩道预备情况,预备后根管深度。
桩道消毒与清洁:根管消毒剂名称、消毒步骤。
试放纤维桩:桩体和桩道密和性、是否裁剪纤维桩、是否使用辅桩等。
粘接纤维桩:粘接剂系统名称类型、粘接过程。
制作树脂核:分层堆核、固化。
牙体预备:肩台位置、边缘形态、冠修复体类型。
制取印模:印模材料的名称、型号。
③修复后评估
依据临床评价标准中有关的项目对修复体进行评价,拍摄根尖片,并记录评价的结果。
④临床观察时间
医疗器械产品根据牙科纤维桩产品特点确定合适的临床观察及随访时间。建议临床试验观察时间点为佩戴桩核冠修复体后1周、3个月、6个月。属于下列情形的建议适当延长临床试验观察时间,如:A.牙科纤维桩产品组成材料缺乏临床应用的安全数据。B.缺乏申报产品临床观察期内(6个月)各性能趋于稳定的数据。C.应用技术与已上市的牙科纤维桩产品不同。
(5)样本量估计
可根据以下六方面确定样本量估计的参数,即:
①拟采取的试验设计类型;
②拟采取的比较类型:优效检验、等效或非劣效检验;
③允许犯假阳性错误的概率a(通常不超过双侧0.05)和犯假阴性错误的概率b(通常不超过0.2);
④主要评价指标的类型和有关的效应大小及其变异程度;
⑤如果是等效或非劣效检验,应提供有临床意义的界值;
⑥根据实际情况,确定可能的病例脱落率,以保证有足够的把握度检测组间差异。
(6)统计分析
应根据试验设计类型、比较类型、资料性质和统计分析目的,合理选择统计分析方法,明确交代统计分析数据集定义及统计分析软件。
统计分析内容应至少包括如下四部分:
①临床试验完成情况描述:包括临床试验概况(筛选人数、入组人数、完成人数、失访/退出/剔除人数等);
②基线描述:应对所有入选受试者(ITT分析集)的基线人口统计学指标及其他相关病史指标等进行统计描述;
③疗效/效果评价:应对所有入选的受试者(ITT分析集)和最终完成试验的受试者(PP分析集)分别进行统计分析,以评价结果的一致性。疗效分析时,除点估计外,还应给出点估计的95%的置信区间估计;
④安全性评价时,应对所有入选的受试者进行分析(SS分析集),不能遗漏所有发生的任何不良事件,对所有发生的不良事件应评价其是否与所研究产品有关。
(7)临床试验报告
临床试验报告应与临床试验方案保持一致,尤其注意明确以下内容:试验产品的名称;规格型号;修复体类型;修复体应用部位;各个病种的病例数;各病例的随访时间;试验产品的临床适应症、禁忌症与注意事项。
临床试验报告中需明确所有病例是否全部完成随访,完成随访病例是否均纳入统计,失访病例需明确失访原因。
临床试验报告中需提交参与疗效评价与安全性评价的统计过程中所涉及到的原始数据。
临床试验报告中需报告所有不良反应和不良事件发生的时间、发生的原因、结果及与试验用产品的关系。对于所采取的措施需予以明确。
临床试验报告应由研究单位根据统计分析报告,出具明确的临床试验结论。
深圳鸿远医疗器械咨询有限公司是一家技术专业的医疗器械咨询服务公司,专注提供全国各地如:深圳、广州、东莞、中山、佛山、潮州、顺德、上海、西安、重庆、成都等知名城市的医疗器械领域技术咨询服务。专业的医疗器械产品注册证咨询代办理、医疗器械产品分类界定代办理、医疗器械生产许可证、三类医疗器械经营许可证、二类医疗器械经营备案、进口医疗器械注册、一类医疗器械产品备案及生产备案、FDA注册、ISO13485认证、 CE认证、计量器具生产许可证、免临床试验、出口证,自由销售证等代办理、医疗器械质量管理体系认证文件的建立及体系与过程确认文件的建立 (如:ISO9001、 ISO13485 、GMP、 CE、QSR820、CMDCAS)产品检测,临床试验及免临床资料编写、产品技术要求制订、技术文件编写辅导、电磁兼容整改、医疗器械广告批文申请办理、洁净室建设指导等服务,技术专业,诚信服务,代理费用低,欢迎您咨询!
|

