|
ЎЎЎЎ
ЎЎЎЎТЅБЖЖчРµЧўІбЧЙСЇ¶юАаїµёґСµБ·ґІУГУЪНЁ№эёД±дМеО»ЎўЖрБўЅЗ¶И¶Ф»јХЯЅшРРСµБ·ґЩЅшїµёґµДїµёґСµБ·ґІЈ¬ёГАаІъЖ·НЁіЈє¬УРµз¶ЇїШЦЖЧ°ЦГЈ¬№ЬАнАа±рОЄIIАаЈ¬ІъЖ··ЦАа±аВлОЄ19-02-02ЎЈ
ЎЎЎЎїµёґСµБ·ґІЧўІбЙкЗлЧКБПТЄЗу
ЎЎЎЎЖуТµФЪЙкЗлїµёґСµБ·ґІЧўІбЦ¤К±РиТЄёщѕЭЎ¶їµёґСµБ·ґІЧўІбЙуІйЦёµјФФтЎ·МбЅ»ПаУ¦ЧўІбЧКБПЈ¬ѕЯМеЧКБПДїВјј°ТЄЗуИзПВЈє
ЎЎЎЎ(Т»)ја№ЬРЕПў
ЎЎЎЎ1.ІъЖ·ГыіЖ
ЎЎЎЎІъЖ·ГыіЖУ¦·ыєПЎ¶ТЅБЖЖчРµНЁУГГыіЖГьГы№жФтЎ·Ў¶ТЅБЖЖчРµНЁУГГыіЖГьГыЦёµјФФтЎ·Ў¶ТЅУГїµёґЖчРµНЁУГГыіЖГьГыЦёµјФФтЎ·µДТЄЗуЎЈ
ЎЎЎЎІъЖ·ГыіЖКѕАэЈєЦ±БўїµёґСµБ·ґІЎўµз¶ЇЖрБўґІЎў¶аМеО»їµёґСµБ·ґІЎЈ
ЎЎЎЎ2.№ЬАнАа±рєН·ЦАа±аВл
ЎЎЎЎёГАаІъЖ·ФЪЎ¶ТЅБЖЖчРµ·ЦАаДїВјЎ·ЦР№ЬАнАа±рОЄIIАаЈ¬·ЦАа±аВлОЄ19-02-02ЎЈ
ЎЎЎЎ3.ЧўІбµҐФЄ»®·Ц
ЎЎЎЎТЅБЖЖчРµЧўІбЙк±ЁІъЖ·ЧўІбµҐФЄ»®·ЦУ¦°ґХХЎ¶ТЅБЖЖчРµЧўІбУл±ё°ё№ЬАн°м·ЁЎ·Ў¶ТЅБЖЖчРµЧўІбµҐФЄ»®·ЦЦёµјФФтЎ·ТЄЗуЈ¬ТФІъЖ·µДјјКхФАнЎўЅб№№ЧйіЙЎўРФДЬЦё±кЎўККУГ·¶О§µИОЄ»®·ЦТАѕЭЎЈ
ЎЎЎЎ3.1ІъЖ·Ѕб№№ЧйіЙј°ЖдМШµгґжФЪЅПґуІоТмµДЈ¬У¦»®·ЦОЄІ»Н¬µДЧўІбµҐФЄЈ¬ИзЦ±БўїµёґСµБ·ґІЎў¶аМеО»їµёґСµБ·ґІЎўPTїµёґСµБ·ґІУ¦»®·ЦОЄІ»Н¬ЧўІбµҐФЄЎЈ(“PT”µДє¬ТеЈєіцґ¦АґФґУЪ±кЧјGB/T 26340Ј¬“P”Ўў“T”·Ц±рОЄ“ОпАн”Ўў“ЦОБЖ”µДУўОДКЧЧЦДёЎЈ)
ЎЎЎЎ3.2ґ«¶Ї·ЅКЅІ»Н¬µДІъЖ·У¦»®·ЦОЄІ»Н¬µДЧўІбµҐФЄЈ¬ИзЈєµз¶ЇТєС№їµёґСµБ·ґІєНµз¶Ї»ъРµїµёґСµБ·ґІІ»ДЬ»®·ЦФЪН¬Т»ЧўІбµҐФЄДЪЎЈ
ЎЎЎЎ3.3µз»ч·А»¤АаРНІ»Н¬µДУ¦»®·ЦОЄІ»Н¬ЧўІбµҐФЄЈ¬Изµз»ч·А»¤АаРНОЄIАаєНµз»ч·А»¤АаРНОЄIIАаµДУ¦»®·ЦОЄІ»Н¬ЧўІбµҐФЄЎЈ
ЎЎЎЎ(¶ю)ТЅБЖЖчРµІъЖ·ЧўІбЧЫКцЧКБП
ЎЎЎЎ1.ІъЖ·ГиКц
ЎЎЎЎ1.1ЖчРµј°ІЩЧчФАнГиКц
ЎЎЎЎ1.1.1№¤ЧчФАн
ЎЎЎЎЦ±БўїµёґСµБ·ґІКЗНЁ№эµчХыЗгР±ЅЗ¶ИК№±»ёїУЪЖдЙПµД»јХЯІъЙъЧФЙнЦШБ¦ЧчУГЈ¬°пЦъ»јХЯНкіЙСцОФО»µЅХѕБўО»Ј¬ЦШРДґУµНµЅёЯµД№э¶ЙЈ¬К№»јХЯід·ЦККУ¦БўО»ЧґМ¬ЎЈМбёЯЗыёЙєНПВЦ«µДёєЦШДЬБ¦Ј¬ФцјУѕ±ЎўРШЎўСьј°№ЗЕиФЪБўО»ЧґМ¬ПВµДїШЦЖДЬБ¦Ј¬ОЄЅ«АґµДЧФЦчБўО»ј°ЖЅєвµД±ЈіЦґтПВБјєГµД»щґЎЎЈ
ЎЎЎЎ¶аМеО»їµёґСµБ·ґІУРБЅїйТФЙПґІ°еЈ¬НЁ№эїШЦЖЧ°ЦГїЙТФ¶АБўµчЅЪёчґІГжµДёЯ¶ИєНЅЗ¶ИЈ¬НЁ№э»ъРµЦ§іЕПµНіЦ§іЕСµБ·ґІЧФЙнТФј°ґІЙП»јХЯµДЙэЅµЈ¬ґЩК№ґІМеёЯ¶Иј°ґІГжёч¶ОО»І»Н¬ЅЗ¶ИµДµчЅЪµГµЅјт»ЇЎЈСµБ·О»ЦГДЬµГµЅід·ЦµДµчХыЈ¬ёЁЦъК№УГХЯЅшРР¶аЦЦЧЛКЖСµБ·ЎЈ
ЎЎЎЎPTїµёґСµБ·ґІКЗІЙУГµз»ъЎўїШЦЖєРј°ЅЕїШїЄ№Ш(ИзККУГ)µИЅшРРµчЅЪїШЦЖµДїµёґСµБ·ґІЎЈїЙёщѕЭїµёґ»јХЯµДРиТЄЈ¬¶ФґІГжёЯ¶ИЎўЅЗ¶И(БЅ¶ОґІ°еККУГ)ЅшРРµчЅЪЈ¬»јХЯЕдєПЦОБЖК¦ЅшРРМеО»µчХыЈ¬·Ѕ±гЦОБЖК¦¶Ф»јХЯИ«ЙнІїО»ЅшРРХп¶ПЎўјмІйЎўЦОБЖєН°ґД¦ЎЈ
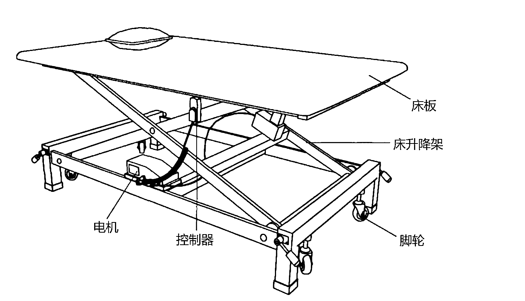
ЎЎЎЎ1.1.2Ѕб№№ЧйіЙ
ЎЎЎЎУ¦ПкПёГиКцІъЖ·Ѕб№№ЧйіЙЎўЦчТЄ№¦ДЬј°ЖдЧйіЙІїјю(№ШјьЧйјюєНИнјю)µД№¦ДЬЎўІъЖ·НјКѕ(є¬ЅУїЪЎўІЩїШГж°еЎўУ¦УГІї·ЦµИПёЅЪ)Ј¬є¬УР¶аёцЧйіЙІї·ЦµДЈ¬У¦ЛµГчЖдБ¬ЅУ·ЅКЅ»тЧйЧ°№ШПµЎЈ
ЎЎЎЎЦ±БўїµёґСµБ·ґІЈєґІГжїЙТФµчЅЪЖрБўЅЗ¶ИЈ¬ёЁЦъЦ§іЕК№УГХЯЅшРРХѕБўСµБ·µДґІЎЈ
ЎЎЎЎЦ±БўїµёґСµБ·ґІНЁіЈУЙґІ°еЎў»ъРµЦ§іЕІїјюЎўµз¶ЇїШЦЖЧ°ЦГЎў№М¶Ё±Ј»¤Ч°ЦГЎў·цКЦЎўЅЕВЦЎўЅЕНР°еµИЧйіЙЎЈ
ЎЎЎЎ¶аМеО»їµёґСµБ·ґІЈєУРБЅїйТФЙПґІ°еЈ¬їЙТФ¶АБўµчЅЪґІГжµДёЯ¶ИєН(»т)ЅЗ¶ИЈ¬ёЁЦъК№УГХЯЅшРР¶аЦЦЧЛКЖСµБ·ЎЈ
ЎЎЎЎ¶аМеО»їµёґСµБ·ґІНЁіЈУЙБЅ¶ОТФЙПґІ°еЎў»ъРµЦ§іЕІїјюЎўµз¶ЇїШЦЖЧ°ЦГЎўЅЕВЦµИЧйіЙЎЈ
ЎЎЎЎPTїµёґСµБ·ґІЈєФЪОпАнЦОБЖК¦ёЁЦъЗыМеФЛ¶ЇСµБ·К±К№УГµДЈ¬їЙТФµчЅЪёЯ¶ИЎўЅЗ¶И(БЅ¶ОґІ°еККУГ)µДґІЎЈ
ЎЎЎЎPTїµёґСµБ·ґІНЁіЈУЙТ»¶О»тБЅ¶ОґІ°еЎў»ъРµЦ§іЕІїјюЎўµз¶ЇїШЦЖЧ°ЦГЎўЅЕВЦµИЧйіЙЎЈ
ЎЎЎЎ1.2РНєЕ№жёс
ЎЎЎЎ¶ФУЪґжФЪ¶аЦЦРНєЕ№жёсµДІъЖ·Ј¬У¦µ±ГчИ·ёчРНєЕ№жёсµДЗш±рЎЈУ¦µ±ІЙУГ¶Ф±И±н»тґшУРЛµГчРФОДЧЦµДНјЖ¬ЎўНј±нЈ¬ГиКцёчЦЦРНєЕ№жёсµДЅб№№ЧйіЙ(»тЕдЦГ)Ўў№¦ДЬЎўІъЖ·МШХчєНФЛРРДЈКЅЎўјјКхІОКэµИДЪИЭЎЈ
ЎЎЎЎ1.3°ьЧ°ЛµГч
ЎЎЎЎУ¦µ±ГиКцІъЖ·°ьЧ°µДАаРНЎўІДЦКµИЈ¬ТФј°УлёГІъЖ·Т»ЖрПъКЫµДЕдјю°ьЧ°ЗйїцЈ¬°ьАЁФЛКд°ьЧ°µДРЕПўЎЈ
ЎЎЎЎ1.4СР·ўАъіМ
ЎЎЎЎИфґжФЪїЙТФІОїјµДН¬АаІъЖ·»тЗ°ґъІъЖ·Ј¬У¦ІыКцЙкЗлЧўІбТЅБЖЖчРµµДІъЖ·їЄ·ўµД±іѕ°єНДїµДЎЈ¶ФУЪН¬АаІъЖ·Ј¬У¦ЛµГчСЎФсЖдЧчОЄСРѕїїЄ·ўЛщІОїјµДФТтЎЈУ¦БР±н±ИЅПЛµГчЙк±ЁІъЖ·УлН¬АаІъЖ·»тЗ°ґъІъЖ·ФЪ№¤ЧчФАнЎўЅб№№ЧйіЙЎўРФДЬЦё±кЎўККУГ·¶О§µИ·ЅГжµДТмН¬Ј¬ІўЦШµгЛµГчЙк±ЁІъЖ·µДРВ№¦ДЬЎўРВУ¦УГЎўРВМШХчЎЈ
ЎЎЎЎ2.ККУГ·¶О§єНЅыјЙЦ¤
ЎЎЎЎ2.1ККУГ·¶О§
ЎЎЎЎУГУЪ¶ФДФЦР·зЎўДФНвЙЛµИ»јХЯЅшРРЦ«МеФЛ¶ЇїµёґСµБ·ЎЈ
ЎЎЎЎ2.2Ф¤ЖЪК№УГ»·ѕі
ЎЎЎЎёГІъЖ·Ф¤ЖЪК№УГµДµШµгИзТЅБЖ»ъ№№Ј¬ТФј°їЙДЬ»бУ°ПмЖд°ІИ«РФєНУРР§РФµД»·ѕіМхјю(ИзЈ¬ОВ¶ИЎўКЄ¶ИЎў№¦ВКЎўС№Б¦ЎўТЖ¶ЇЎўµзФґМхјюµИ)ЎЈ
ЎЎЎЎ2.3ККУГИЛИє
ЎЎЎЎДї±к»јХЯИЛИєµДРЕПў(ИзіЙИЛЎў¶щНЇ)Ј¬»јХЯСЎФс±кЧјµДРЕПўЈ¬ТФј°К№УГ№эіМЦРРиТЄјаІвµДІОКэЎўїјВЗµДТтЛШЎЈ
ЎЎЎЎ2.4ЅыјЙЦ¤
ЎЎЎЎИзККУГЈ¬У¦№ШЧўКЗ·сТСГчИ·ЛµГчёГЖчРµІ»ККТЛУ¦УГµДДіР©јІІЎЎўЗйїц»тМШ¶ЁµДИЛИє(Из¶щНЇЎўАПДкИЛЎўФРёѕј°ІёИйЖЪёѕЕ®ЎўёОЙц№¦ДЬІ»И«ХЯ)ЎЈ
ЎЎЎЎ(Иэ)·ЗБЩґІЧКБП
ЎЎЎЎ1.ІъЖ··зПХ№ЬАнЧКБП
ЎЎЎЎТЅБЖЖчРµЧўІб°мАнЙкЗлИЛУ¦ІОХХGB/T 42062µД№ж¶ЁЈ¬ІўЅбєПІъЖ·МШµг¶ФІъЖ··зПХЅшРРИ«ЙъГьЦЬЖЪµД№ЬАнЎЈ·зПХ№ЬАн»о¶ЇТЄ№бґ©ІъЖ·ЙијЖЎўЙъІъЎўЙПКРєуК№УГј°ІъЖ·ґ¦АнµДХыёцЙъГьЦЬЖЪЎЈ·зПХ№ЬАн±ЁёжїЙ°ьє¬·зПХ·ЦОцЎў·зПХЖАјЫЎў·зПХїШЦЖЎў·зПХјаІвЈ¬У¦·ыєПЎ¶ТЅБЖЖчРµ·зПХ№ЬАн¶ФТЅБЖЖчРµµДУ¦УГЎ·(GB/T 42062)µДУР№ШТЄЗуЈ¬ЙуІйТЄµг°ьАЁЈє
ЎЎЎЎ1.1КЗ·сХэИ·К¶±рУлІъЖ·°ІИ«УР№ШµДМШХчЎЈ
ЎЎЎЎ1.2КЗ·сПµНіК¶±рХэіЈєН№КХПБЅЦЦМхјюПВµДїЙФ¤јыОЈПХ(Фґ)ЎЈ
ЎЎЎЎ1.3КЗ·сАыУГ·зПХ№ЬАнјЖ»®ЦР№ж¶ЁµДїЙЅУКЬЧјФтЈ¬¶Ф·зПХЅшРРЖАјЫІўЅшРР·зПХїШЦЖЈ¬ТІ°ьАЁЧЫєПКЈУа·зПХµДїЙЅУКЬРФЖАјЫј°ЙъІъєНЙъІъєујаКУПа№Ш·Ѕ·ЁЎЈ
ЎЎЎЎёЅјю6-2ёшіцБЛїµёґСµБ·ґІіЈјыµД·зПХТЄЛШј°КѕАэЎЈУЙУЪІъЖ·µД№¤ЧчФАнЎўЅб№№ЧйіЙЎўРФДЬЦё±кµИґжФЪІоТмЈ¬ЛщТФХвР©·зПХТЄЛШІўІ»КЗИ«ІїЈ¬ЧўІбЙкЗлИЛУ¦°ґХХGB/T 42062Ў¶ТЅБЖЖчРµ·зПХ№ЬАн¶ФТЅБЖЖчРµµДУ¦УГЎ·ЦР№ж¶ЁµД№эіМєН·Ѕ·ЁЈ¬ФЪІъЖ·ХыёцЙъГьЦЬЖЪДЪЅЁБўЎўРОіЙОДјюІў±ЈіЦТ»ёціЦРшµД№эіМЈ¬УГТФЕР¶ЁУлТЅБЖЖчРµУР№ШµДОЈПХ(Фґ)Ўў№АјЖєНЖАјЫПа№ШµД·зПХЎўїШЦЖХвР©·зПХІўјаКУЙПКцїШЦЖµДУРР§РФЈ¬ТФід·Ц±ЈЦ¤ІъЖ·µД°ІИ«єНУРР§ЎЈ
ЎЎЎЎ2.ТЅБЖЖчРµ°ІИ«єНРФДЬ»щ±ѕФФтЗ嵥
ЎЎЎЎЛµГчІъЖ··ыєПЎ¶ТЅБЖЖчРµ°ІИ«єНРФДЬ»щ±ѕФФтЗ嵥Ў·ёчПоККУГТЄЗуЛщІЙУГµД·Ѕ·ЁЈ¬ТФј°Ц¤ГчЖд·ыєПРФµДОДјюЎЈ¶ФУЪЎ¶ТЅБЖЖчРµ°ІИ«єНРФДЬ»щ±ѕФФтЗ嵥Ў·ЦРІ»ККУГµДёчПоТЄЗуЈ¬У¦µ±ЛµГчАнУЙЎЈ
ЎЎЎЎ¶ФУЪ°ьє¬ФЪІъЖ·ЧўІбЙк±ЁЧКБПЦРµДОДјюЈ¬У¦µ±ЛµГчЖдФЪЙк±ЁЧКБПЦРµДѕЯМеО»ЦГ;¶ФУЪОґ°ьє¬ФЪІъЖ·ЧўІбЙк±ЁЧКБПЦРµДОДјюЈ¬У¦µ±ЧўГчёГЦ¤ѕЭОДјюГыіЖј°ЖдФЪЦКБї№ЬАнМеПµОДјюЦРµД±аєЕ±ёІйЎЈ
ЎЎЎЎ3.ТЅБЖЖчРµЧўІбЦ¤ЙкЗлІъЖ·јјКхТЄЗуј°јмСй±Ёёж
ЎЎЎЎ3.1јјКхТЄЗу
ЎЎЎЎјјКхТЄЗуУ¦µ±°ґХХЎ¶ТЅБЖЖчРµІъЖ·јјКхТЄЗу±аРґЦёµјФФтЎ·µД№ж¶Ё±аЦЖЎЈїЙТФёщѕЭІъЖ·µДјјКхМШµгЦЖ¶ЁПаУ¦µДјјКхТЄЗ󣬵«РФДЬЦё±кІ»µГµНУЪПа№Ш№ъјТ±кЧјЎўРРТµ±кЧјµДУР№ШТЄЗуЈ¬ИзУРІ»ККУГМхїоЈ¬У¦ЛµГчАнУЙЎЈ
ЎЎЎЎІъЖ·јјКхТЄЗуГчИ·ІъЖ·№жёсРНєЕј°»®·ЦЛµГчЎЈґжФЪ¶аЦЦРНєЕµДЈ¬У¦ГчИ·І»Н¬РНєЕЦ®јдµДТмН¬ЎЈИфє¬УРИнјюЧйјюЈ¬У¦ТЄЧўГчИнјю·ўІј°ж±ѕЎўИнјю°ж±ѕГьГы№жФтЈ¬ЖдЦРИнјю°ж±ѕГьГы№жФтРиУлЦКБї№ЬАнМеПµ±ЈіЦТ»ЦВЎЈ
ЎЎЎЎ3.1.1РФДЬЦё±к
ЎЎЎЎ3.1.1.1їµёґСµБ·ґІУ¦µ±·ыєПGB/T 26340Ў¶їЙµчКЅїµёґСµБ·ґІЎ·µДТЄЗуЎЈ
ЎЎЎЎ3.1.1.2ЕдУРЅЕМ¤їЄ№ШµДЙи±ёЈ¬У¦·ыєПYY/T 1057Ў¶ТЅУГЅЕМ¤їЄ№ШНЁУГјјКхМхјюЎ·µДТЄЗуЎЈ
ЎЎЎЎ3.1.1.3ИнјюЈєИфККУГЈ¬У¦°ґХХЎ¶ТЅБЖЖчРµИнјюЧўІбјјКхЙуІйЦёµјФФтЎ·(2022ДкРЮ¶©°ж)БРГчИнјюРФДЬЦё±кЎЈ“РФДЬЦё±к”°ьАЁИнјюµД№¦ДЬЎўК№УГПЮЦЖЎўЅУїЪЎў·ГОКїШЦЖЎўФЛРР»·ѕі(ИфККУГ)ЎўРФДЬР§ВК(ИфККУГ)µИТЄЗуЎЈ
ЎЎЎЎ3.1.2µзЖш°ІИ«ТЄЗу
ЎЎЎЎµзЖш°ІИ«У¦µ±·ыєПGB 9706.1Ў¶ТЅУГµзЖшЙи±ё µЪ1Ії·ЦЈє»щ±ѕ°ІИ«єН»щ±ѕРФДЬµДНЁУГТЄЗуЎ·ЎўGB 24436Ў¶їµёґСµБ·ЖчРµ°ІИ«НЁУГТЄЗуЎ·(5.10µзЖч°ІИ«іэНв)єНGB/T 26340Ў¶їЙµчКЅїµёґСµБ·ґІЎ·ЦРµЪ7МхµДТЄЗуЎЈ
ЎЎЎЎ3.1.3µзґЕјжИЭТЄЗу
ЎЎЎЎµзґЕјжИЭУ¦·ыєПYY 9706.102Ў¶ТЅУГµзЖшЙи±ё µЪ1-2Ії·ЦЈє»щ±ѕ°ІИ«єН»щ±ѕРФДЬµДНЁУГТЄЗуІўБР±кЧјЈєµзґЕјжИЭ ТЄЗуєНКФСйЎ·µДТЄЗуЎЈ
ЎЎЎЎ3.2ІъЖ·јмСй±Ёёж
ЎЎЎЎТЅБЖЖчРµІъЖ·ЧўІбЦ¤ЙкЗлИЛУ¦°ґХХІъЖ·јјКхТЄЗуЅшРРјмСйЈ¬ІўМбЅ»јмСй±ЁёжЎЈУ¦ЛµГчјмСйУГРНєЕ№жёсµДµдРНРФЎЈ
ЎЎЎЎµдРНРНєЕІъЖ·ФФтЙПУ¦КЗН¬Т»ЧўІбµҐФЄДЪДЬ№»ґъ±н±ѕµҐФЄДЪЖдЛыІъЖ·°ІИ«РФєНУРР§РФµДІъЖ·Ј¬У¦їјВЗ№¦ДЬЧоЖлИ«ЎўЅб№№ЧйіЙЧоёґФУЎў·зПХЧоёЯµДІъЖ·ЎЈ
ЎЎЎЎ4.СРѕїЧКБП
ЎЎЎЎ4.1»ЇС§єНОпАнРФДЬСРѕї
ЎЎЎЎУ¦µ±МṩІъЖ·»ЇС§/ІДБП±нХчЎўОпАнєН/»т»ъРµРФДЬЦё±кµДИ·¶ЁТАѕЭЎўЙијЖКдИлАґФґТФј°БЩґІТвТеЈ¬ЛщІЙУГµД±кЧј»т·Ѕ·ЁЎўІЙУГµДФТтј°АнВЫ»щґЎЎЈ
ЎЎЎЎ№¦ДЬРФЦё±кµДСйЦ¤У¦ёщѕЭЧЫКцЧКБПЦРУР№ШЙк±ЁІъЖ·Ѕб№№ЧйіЙµДЗйїцЈ¬їЙІОїјGB/T 26340µДТЄЗуЈ¬ЦБЙЩУ¦°ьАЁТФПВЦё±кЈє»о¶ЇІїјюЎўГж±ЯЅЗєН№Ь¶ЛЎўЅб№№ЙијЖЎўОИ¶ЁРФЎў»ъРµЗї¶ИЎўПЯРФіЯґзУлЅЗ¶ИТЄЗуЎўІЩЧчБ¦ЎўІЩЧчЛЩ¶ИєНК±јдЎўЅЕВЦ¶ЁО»ЎўФлТфЎўїШЦЖЖчЎўІЩЧчКЦ±ъєНМ¤°еЎўµзФґИнµзПЯµИЎЈЦ±БўїµёґСµБ·ґІ»№У¦°ьАЁёЅјюј°Зї¶ИТЄЗуЈ¬ёЅјУОИ¶ЁРФТЄЗуЎЈ
ЎЎЎЎ4.2µзЖш°ІИ«РФСРѕї
ЎЎЎЎМṩµзЖш°ІИ«РФЎў»ъРµєН»·ѕі±Ј»¤ТФј°µзґЕјжИЭРФµДСРѕїЧКБПЈ¬ЛµГчККУГµД±кЧјТФј°їЄХ№µДСРѕїЎЈ°ІИ«Цё±кУ¦µ±°ьАЁGB 9706.1ЎўGB 24436(5.10 µзЖч°ІИ«МхїоіэНв)ј°ЖдЛыККУГµД№ъјТ±кЧјєНРРТµ±кЧјЦРµДЛщУРЦё±кЈ¬µзґЕјжИЭЦё±кУ¦µ±°ьАЁYY 9706.102ј°ЖдЛыККУГµД№ъјТ±кЧјєНРРТµ±кЧјЦРµДЛщУРЦё±кЎЈ
ЎЎЎЎ4.3ИнјюСРѕї
ЎЎЎЎУ¦№ШЧўКЗ·сТС°ґХХЎ¶ТЅБЖЖчРµИнјюЧўІбЙуІйЦёµјФФтЎ·(2022ДкРЮ¶©°ж)µДТЄЗуМбЅ»ИнјюПа№ШЧКБПЎЈИзОЄЧФСРИнјюЈ¬У¦МṩЧФСРИнјюСРѕї±ЁёжЎЈМбЅ»ЧФСРИнјюСРѕї±ЁёжЎўНвІїИнјю»·ѕіЖА№А±Ёёж(ИфККУГ)ТФј°GB/T 25000.51ЧФІв±ЁёжЈ¬ТаїЙМбЅ»ЧФјм±Ёёж»тјмСй±ЁёжґъМжЧФІв±ЁёжЎЈИзОЄПЦіЙИнјюЈ¬У¦МṩПЦіЙИнјюЧйјюСРѕїЧКБПЎЈ
ЎЎЎЎТЅБЖЖчРµЧўІбЦ¤ґъАнИзЙк±ЁІъЖ·ККУГУЪТЅБЖЖчРµНшВз°ІИ«Ј¬°ьАЁѕЯ±ёµзЧУКэѕЭЅ»»»ЎўФ¶іМ·ГОКУлїШЦЖЎўУГ»§·ГОКИэЦЦ№¦ДЬµ±ЦРТ»ЦЦј°ТФЙП№¦ДЬЈ¬У¦°ґХХЎ¶ТЅБЖЖчРµНшВз°ІИ«ЧўІбЙуІйЦёµјФФтЎ·(2022ДкРЮ¶©°ж)Ј¬МбЅ»ТЅБЖЖчРµНшВз°ІИ«СРѕїЧКБПЎЈ
ЎЎЎЎ4.4ЙъОпС§МШРФСРѕї
ЎЎЎЎЦ±БўїµёґСµБ·ґІµДґІГжЎў°уґшЎўЧА°е»т·цКЦЈ¬¶аМеО»їµёґСµБ·ґІЎўPTїµёґСµБ·ґІµДґІГжµИІїјюїЙДЬ»бУл»јХЯµД±нГжНкєГЖ¤·фґжФЪ¶МЖЪ(≤24h)Ц±ЅУЅУґҐЈ¬У¦ЅшРРЙъОпПаИЭРФЖАјЫЎЈЙъОпПаИЭРФЖАјЫїЙёщѕЭGB/T 16886.1єНЎ¶№ШУЪУЎ·ўТЅБЖЖчРµЙъОпС§ЖАјЫєНЙуЖАЦёДПµДНЁЦЄЎ·µДТЄЗуЅшРРЎЈУ¦ЖАјЫµДПоДїЦБЙЩ°ьАЁЈєПё°ы¶ѕРФЎўЦВГф·ґУ¦ЎЈ
ЎЎЎЎИфЧўІбЙкЗлИЛДЬ№»Ц¤ГчЦЖФмЙПКцІїјюЛщУГІДБПТСѕИ·БўБЛ°ІИ«К№УГК·Ј¬ФтїЙІ»ФЩїЄХ№ЙъОпПаИЭРФЖАјЫЎЈВЫЦ¤ІДБПµД°ІИ«К№УГК·К±Ј¬У¦ДЬ№»ЛµГчЦЖФмЙПКцІїјюК±ЛщУГµДФІДБПЎў»ЇС§Оп(ЦъјБЎўМнјУјБµИ)єНјУ№¤№эіМ;У¦ДЬ№»МṩЙПКцІїјюФЪН¬µИЅУґҐМхјю»тёьОЄ¶сБУЅУґҐМхјюПВµДУ¦УГЗйїцЈ¬»тДЬ№»МṩЙПКцІїјюТСѕїЄХ№µД·ыєПЙъОпПаИЭРФТЄЗуµДЖАјЫЧКБПЎЈИфЙПКцІїјюОЄНв№єІїјюЈ¬їЙИПїЙєПёс№©·ЅіцѕЯЦ¤ГчЧКБПЈ¬Ц¤ГчЖдТСѕИ·БўБЛ°ІИ«К№УГК·ЎЈ(ЧўЈєє¬УРОґѕК№УГµДРВІДБПЎў»ЇС§Оп»тјУ№¤№эіМµДІїјюІ»ККУГУЪ±ѕМхїоЎЈ)
ЎЎЎЎЙъОпПаИЭРФЖАјЫСРѕїЧКБПУ¦µ±°ьАЁЈєЙъОпПаИЭРФЖАјЫµДТАѕЭєН·Ѕ·Ё;ІъЖ·ЛщУГІДБПµДГиКцј°УлИЛМеЅУґҐµДРФЦК;КµК©»т»нГвЙъОпС§КФСйµДАнУЙєНВЫЦ¤;¶ФУЪПЦУРКэѕЭ»тКФСйЅб№ыµДЖАјЫЎЈ
ЎЎЎЎ4.5ЗеЅај°Пы¶ѕ№¤ТХСРѕї
ЎЎЎЎК№УГХЯЗеЅаєНПы¶ѕЈєУ¦µ±ГчИ·НЖјцµДЗеПґєНПы¶ѕ№¤ТХ(·Ѕ·ЁєНІОКэ)Ўў№¤ТХµДИ·¶ЁТАѕЭТФј°СйЦ¤µДПа№ШСРѕїЧКБПЎЈ
ЎЎЎЎІРБф¶ѕРФЈєИфІъЖ·ѕПы¶ѕєуїЙДЬІъЙъІРБфОпЦК,У¦µ±¶ФПы¶ѕєуµДІъЖ·ЅшРРІРБф¶ѕРФµДСРѕїЈ¬ГчИ·ІРБфОпРЕПўј°ІЙИЎµДґ¦Ан·Ѕ·ЁЈ¬ІўМṩПа№ШСРѕїЧКБПЎЈ
ЎЎЎЎ5.ОИ¶ЁРФСРѕї
ЎЎЎЎТЅБЖЖчРµЧўІбЦ¤°мАнЙкЗлИЛМṩІъЖ·µДК№УГОИ¶ЁРФЎўїЙїїРФСРѕїЧКБПЈ¬Ц¤ГчФЪ№ж¶ЁµДК№УГЖЪПЮДЪЈ¬ФЪХэіЈК№УГЎўО¬»¤єНРЈЧј(ИзККУГ)ЗйїцПВЈ¬ІъЖ·µДРФДЬ№¦ДЬїЙТФВъЧгБЩґІК№УГТЄЗуЎЈІОїјЎ¶УРФґТЅБЖЖчРµК№УГЖЪПЮјјКхЙуІйЦёµјФФтЎ·µДТЄЗуїЄХ№СРѕїЎЈ
ЎЎЎЎИфТАѕЭ·ЦОц№ШјьІїјюКЩГьАґИ·¶ЁІъЖ·К№УГЖЪПЮЈ¬№ШјьІїјюЦБЙЩУ¦°ьАЁµз»ъЎўїШЦЖЖчµИЎЈІъЖ·ИфѕЯУРїЙёь»»ІїјюЈ¬У¦ГчИ·¶ЁЖЪ±ЈСшО¬»¤К±јдєНёь»»ЖµґОЈ¬ЗТУ¦ёшіцЦ§іЦРФЧКБПЎЈ
ЎЎЎЎМṩІъЖ·µДФЛКдОИ¶ЁРФєН°ьЧ°СРѕїЧКБПЈ¬Ц¤ГчФЪ№ж¶ЁµДФЛКдМхјюПВЈ¬ФЛКд№эіМЦРµД»·ѕіМхјю(АэИзЈєХр¶ЇЎўХс¶ЇЎўОВ¶ИєНКЄ¶ИµДІЁ¶Ї)І»»б¶ФІъЖ·µДМШРФєНРФДЬЈ¬°ьАЁНкХыРФєНЗеЅа¶ИЈ¬ФміЙІ»АыУ°ПмЎЈ
ЎЎЎЎІОХХGB/ T 14710±кЧјµДТЄЗуј°ІъЖ·К№УГ(ОВ¶ИЎўКЄ¶ИЎўЖшС№ЎўµзФґ)ЎўФЛКдЎўґўґжМхјюїЄХ№»·ѕіКФСйСРѕїЎЈ
ЎЎЎЎ6.ЖдЛыЧКБП
ЎЎЎЎТЅБЖЖчРµЧўІбґъАнЧўІбЙкЗлИЛёщѕЭЎ¶№ъјТТ©јаѕЦ№ШУЪ·ўІјГвУЪБЩґІЖАјЫТЅБЖЖчРµДїВјµДНЁёжЎ·Ј¬“ІъЖ·ГыіЖЈєЦ«МеїµёґСµБ·Йи±ёЈ¬·ЦАа±аВлЈє19-02-02”°ьє¬ФЪГвУЪБЩґІЖАјЫТЅБЖЖчРµДїВјЦРЈ¬°ґХХЎ¶№ъјТТ©јаѕЦ№ШУЪ·ўІјТЅБЖЖчРµБЩґІЖАјЫјјКхЦёµјФФтµИ5ПојјКхЦёµјФФтµДНЁёжЦРёЅјю5Ў¶БРИлГвУЪБЩґІЖАјЫТЅБЖЖчРµДїВјІъЖ·¶Ф±ИЛµГчјјКхЦёµјФФтЎ·µДТЄЗуЈ¬ЧўІбЙкЗлИЛРиМбЅ»Йк±ЁІъЖ·Па№ШРЕПўУлЎ¶ДїВјЎ·ЛщКцДЪИЭµД¶Ф±ИЧКБПєНЙк±ЁІъЖ·УлТС»сЧјѕіДЪЧўІбµДЎ¶ДїВјЎ·ЦРТЅБЖЖчРµµД¶Ф±ИЛµГчЎЈѕЯМеРиМбЅ»µДЧКБПТЄЗуИзПВЈє
ЎЎЎЎ6.1МбЅ»Йк±ЁІъЖ·Па№ШРЕПўУлЎ¶ДїВјЎ·ЛщКцДЪИЭµД¶Ф±ИЧКБП;
ЎЎЎЎ6.1МбЅ»Йк±ЁІъЖ·УлЎ¶ДїВјЎ·ЦРТС»сЧјѕіДЪЧўІбТЅБЖЖчРµµД¶Ф±ИЛµГчЈ¬¶Ф±ИЛµГчУ¦µ±°ьАЁЎ¶Йк±ЁІъЖ·УлДїВјЦРТС»сЧјѕіДЪЧўІбТЅБЖЖчРµ¶Ф±И±нЎ·єНПаУ¦Ц§іЦРФЧКБПЎЈИфѕ¶Ф±ИЈ¬Йк±ЁІъЖ·Ул¶Ф±ИІъЖ·ґжФЪІоТмЈ¬»№У¦МбЅ»ІоТмІї·Ц¶Ф°ІИ«УРР§РФУ°ПмµД·ЦОцСРѕїЧКБПЎЈ¶юХЯµДІоТмІ»У¦ТэЖрІ»Н¬µД°ІИ«УРР§РФОКМвЈ¬јґЙк±ЁІъЖ·ОґіцПЦ¶Ф±ИІъЖ·І»ґжФЪµДЗТїЙДЬТэ·ўЦШґу·зПХєН/»тТэЖрПФЦшУ°ПмУРР§РФµДОКМвЎЈ
ЎЎЎЎТЅБЖЖчРµІъЖ·ЧўІб°мАнМбЅ»µДЙПКцЧКБПУ¦ДЬЦ¤ГчЙк±ЁІъЖ·УлЎ¶ДїВјЎ·ЛщКцµДІъЖ·ѕЯУР»щ±ѕµИН¬РФЎЈИфОЮ·ЁЦ¤ГчЙк±ЁІъЖ·УлЎ¶ДїВјЎ·ЛщКцµДІъЖ·ѕЯУР»щ±ѕµИН¬РФЈ¬ФтУ¦їЄХ№БЩґІЖАјЫЎЈ
ЎЎЎЎ(ЛД)БЩґІЖАјЫЧКБП
ЎЎЎЎёГІъЖ·ТСБРИлЎ¶ГвУЪБЩґІЖАјЫТЅБЖЖчРµДїВјЎ·Ј¬ѕЯМеТЄЗуПкјы“·ЗБЩґІЧКБП”µД“ЖдЛыЧКБП”Ії·ЦЎЈ
ЎЎЎЎ(Ое)ІъЖ·ЛµГчКйєН±кЗ©Сщёе
ЎЎЎЎ№ШЧўїµёґСµБ·ґІІъЖ·µДЛµГчКйєН±кЗ©КЗ·с·ыєПЎ¶ТЅБЖЖчРµЛµГчКйєН±кЗ©№ЬАн№ж¶ЁЎ·(№ъјТКіЖ·Т©Ж·ја¶Ѕ№ЬАнЧЬѕЦБоµЪ6єЕ)ЎўYY/T 0466.1ЎўGB 9706.1ЎўGB 24436ЎўGB/T 26340єНYY 9706.102ЦРµДПа№ШТЄЗуЎЈЛµГчКйЎў±кЗ©µДДЪИЭУ¦µ±ХжКµЎўНкХыЎўїЖС§Ј¬ІўУлІъЖ·МШРФПаТ»ЦВЈ¬ОДЧЦДЪИЭ±ШРлК№УГЦРОДЎЈЛµГчКйЎў±кЗ©ЦРµДОДЧЦЎў·ыєЕЎўНјРОЎў±нёсЎўКэѕЭµИУ¦П໥һЦВЈ¬Іў·ыєППа№Ш±кЧјєН№ж·¶ТЄЗуЎЈ
ЎЎЎЎ1.ЛµГчКйіэ·ыєПЙПКц№ж¶ЁНвЈ¬»№У¦°ьАЁµ«І»ПЮУЪТФПВДЪИЭЈє
ЎЎЎЎ1.1ІъЖ·МШ¶ЁИЛИєК№УГµДЛµГчТФј°КЗ·сРиТЄФЪТЅ»¤ИЛФ±µДја»¤ПВК№УГµДЛµГч;
ЎЎЎЎ1.2ІъЖ·јјКхТЄЗуµДЦчТЄРФДЬЦё±кЦРГчИ·РиТЄФЪЛµГчКйЦРГчКѕµДДЪИЭ;
ЎЎЎЎ1.3НкХыµДІЩЧчЛµГчЈєІъЖ·µДК№УГ»·ѕіМхјю;°ІЧ°ј°µчКФЛµГчЦРУ¦ГчИ·°ІЧ°К±¶ФµШГжµДТЄЗу;ПкПёЛµГчІъЖ·µДѕЯМеК№УГ·Ѕ·Ёј°НјКѕ;
ЎЎЎЎ1.4У¦ГчИ·ЕдјюµДёь»»·Ѕ·Ёј°ЧўТвКВПо;У¦ёшіцИнјю°ІЧ°ЎўЙэј¶µИѕЯМеРЕПў;
ЎЎЎЎ1.5ИфМṩїЙУЙК№УГХЯёь»»»тЦШРВБ¬ЅУµД№М¶ЁЧ°ЦГЈ¬ЛµГчКйЦРУ¦ЛµГчСйЦ¤№М¶ЁЧ°ЦГКЗ·сТС±»їЙїїБ¬ЅУµД·Ѕ·Ё;
ЎЎЎЎ1.6ЧўТвКВПоЦРЦБЙЩУ¦ГчИ·ТміЈЗйїцПВЈ¬К§їШЧґМ¬ПВµДЅфј±ґ¦АнґлК©ЎЈМШКвЗйїцПВ(НЈµзЎўТвНвТЖ¶ЇµИ)µДЧўТвКВПоЈ¬їЙДЬіцПЦµДОуІЩЧчј°їЙДЬФміЙµДЙЛє¦;
ЎЎЎЎ1.7ѕЇёжРЕПўЈєАэИзФЪОЮИ˼໤ЗйїцПВЅыЦ№К№УГµДѕЇёжЈ¬І»µ±ІЩЧчїЙДЬґшАґ°ІИ«Тю»јµДѕЇёж;
ЎЎЎЎ1.8°ІИ«№¤ЧчФШєЙ;
ЎЎЎЎ1.9ХыМеіЯґзєНЦШБїЈ¬°ьАЁЦчМеІї·ЦµДЦШБїЎЈ
ЎЎЎЎ1.10Йк±ЁІъЖ·ёЅјюµДЛµГчЎЈ
ЎЎЎЎ2.К№УГЛµГчКйЙуЖА№ШЧўµгЈє
ЎЎЎЎ2.1У¦µ±МṩДвЙк±Ё·¶О§ДЪЛщУРРНєЕµДЛµГчКйЈ¬У¦ёІёЗЛщЙкЗлµДЛщУРЧйіЙІї·ЦЎЈ
ЎЎЎЎ2.2ІъЖ·ГыіЖЎўРНєЕЎў№жёсЎўЦчТЄРФДЬЎўЅб№№УлЧйіЙУ¦УлІъЖ·јјКхТЄЗуДЪИЭТ»ЦВ;ІъЖ·µДККУГ·¶О§У¦УлЧўІбЙкЗл±нЎўІъЖ·јјКхТЄЗуј°БЩґІЖАјЫЧКБПТ»ЦВЎЈ
ЎЎЎЎ2.3ЧўІбИЛ/ЙъІъЖуТµГыіЖЎўЧЎЛщЎўЙъІъµШЦ·ЎўБЄПµ·ЅКЅј°КЫєу·юОсµҐО»У¦ХжКµУРР§Ј¬ЧўІбИЛ/ЙъІъЖуТµГыіЖєНЧЎЛщУ¦Ул Ў¶УЄТµЦґХХЎ·Т»ЦВ;Ў¶ТЅБЖЖчРµЙъІъЖуТµРнїЙЦ¤Ў·±аєЕЎўТЅБЖЖчРµЧўІбЦ¤Кй±аєЕЎўІъЖ·јјКхТЄЗу±аєЕО»ЦГУ¦Ф¤БфЎЈ
ЎЎЎЎ2.4УлЙк±ЁІъЖ·Т»ЖрК№УГµДЖдЛыТЅБЖЖчРµ»тІ»КфУЪТЅБЖЖчРµµДІъЖ·µДГиКцЈ¬ФЪЛµГчКйЦРУ¦ТЄЗуЛщБ¬ЅУЙи±ёУ¦·ыєППаУ¦µД °ІИ«±кЧјЈ¬ІўТЄЗуУлёГЖчРµБ¬ЅУК№УГЧйіЙµДПµНіЛщУ¦·ыєППаУ¦µД°ІИ«±кЧјЈ¬ј°ЖдЛы±ШТЄµДРЕПўЎЈ
ЎЎЎЎ(Бщ)¶юАаТЅБЖЖчРµЧўІбЙкЗлЦКБї№ЬАнМеПµОДјю
ЎЎЎЎЦКБї№ЬАнМеПµОДјюУ¦·ыєПЎ¶ТЅБЖЖчРµЙъІъЦКБї№ЬАн№ж·¶Ў·µДТЄЗуЈ¬Іў°ґХХ№ъјТТ©Ж·ја¶Ѕ№ЬАнѕЦЎ¶№ШУЪ№«ІјТЅБЖЖчРµЧўІбЙк±ЁЧКБПТЄЗуєНЕъЧјЦ¤ГчОДјюёсКЅµД№«ёжЎ·ТЄЗуМбЅ»ЧКБПЎЈ
ЙоЫЪєиФ¶ТЅБЖЖчРµЧЙСЇУРПЮ№«ЛѕКЗТ»јТјјКхЧЁТµµДТЅБЖЖчРµЧЙСЇ№«ЛѕЈ¬ЧЁЧўМṩȫ№ъёчµШИзЈєЙоЫЪЎў№гЦЭЎў¶«ЭёЎўЦРЙЅЎў·рЙЅЎўі±ЦЭЎўЛіµВЎў№гОчЎўЙПєЈЎўОч°ІЎўЦШЗмЎўіЙ¶јЎўХгЅЎўЅЛХµИЦЄГыіЗКРµДТЅБЖЖчРµБмУтјјКхЧЙСЇ·юОсЎЈєиФ¶ТЅБЖЖчРµЧЙСЇЧЁТµ·юОсУЪЈєТЅБЖЖчРµІъЖ·ЧўІбЦ¤ЎўТЅБЖЖчРµЙъІъРнїЙЦ¤ЎўЅшїЪТЅБЖЖчРµЧўІбЎўТ»АаТЅБЖЖчРµІъЖ·±ё°ёј°ЙъІъ±ё°ёґъ°мЎўТЅБЖЖчРµѕУЄРнїЙЦ¤ґъ°мЎў¶юАаТЅБЖЖчРµѕУЄ±ё°ёЎўТЅБЖЖчРµ·ЦАаЅз¶ЁЎўCEИПЦ¤ЎўISO13485ИПЦ¤ЎўFDAЧўІбЎўБЩґІКФСйЎўТЅБЖЖчРµЦКБї№ЬАнМеПµИПЦ¤ј°МеПµЅЁБўУл№эіМИ·ИПОДјюЅЁБў(ISO9001, ISO13485, GMP, CEЈ¬QSR820Ј¬CMDCAS);ІъЖ·јјКхТЄЗуЦЖ¶©ЎўјјКхОДјюЎўБЩґІКФСйј°ГвБЩґІН¬АаІъЖ·±И¶Ф±ИЧКБП±аРґЎўЧўІбЧКБП±аРґёЁµјЎўµзґЕјжИЭФ¤ІвХыёДЎўТЅБЖЖчРµ№гёжЕъОДЙк±ЁЎўТЅБЖЖчРµіцїЪПъКЫЦ¤Гч°мАнЎўЅаѕ»КТЅЁЙиЦёµјµИМṩһվʽ·юОсЈ¬јјКхЧЁТµЈ¬іПРЕ·юОсЈ¬»¶УДъЧЙСЇЎЈ
ЎЎЎЎ
|

