|
ЎЎЎЎ
ТЅБЖЖчРµЧўІбЧЙСЇТЅУГєфОьµАКЄ»ЇЖчЧўІбёщѕЭЎ¶ТЅУГєфОьµАКЄ»ЇЖчЧўІбЙуІйЦёµјФФтЎ·Йк±ЁЧКБПРиТЄМбЅ»ПВЧКБПЈ¬ІўТФ·ыПа№ШТЄЗуЈє
ЎЎЎЎ(Т»)ја№ЬРЕПў
ЎЎЎЎ1.ІъЖ·ГыіЖ
ЎЎЎЎІъЖ·ГыіЖУ¦ОЄНЁУГГыіЖЈ¬Іў·ыєПЎ¶ТЅБЖЖчРµНЁУГГыіЖГьГы№жФтЎ·µИПа№Ш·Ё№жЎў№ж·¶РФОДјюµДТЄЗуЈ¬Из“ТЅУГєфОьµАКЄ»ЇЖч”µИЎЈ
ЎЎЎЎ2.№ЬАнАа±рєН·ЦАа±аВл
ЎЎЎЎІъЖ·°ґµЪ¶юАаТЅБЖЖчРµ№ЬАнЈ¬·ЦАа±аВлОЄ08-05-02ЎЈ
ЎЎЎЎ3.ТЅБЖЖчРµЧўІбµҐФЄ»®·Ц
ЎЎЎЎЧўІбµҐФЄ°ґХХЎ¶ТЅБЖЖчРµЧўІбµҐФЄ»®·ЦЦёµјФФтЎ·µДТЄЗуЈ¬ФФтЙПТФІъЖ·µДјјКхФАнЎўЅб№№ЧйіЙЎўРФДЬЦё±кєНККУГ·¶О§ОЄ»®·ЦТАѕЭЎЈ
ЎЎЎЎјУИИКЄ»ЇїШЦЖјјКхФАнІо±рЅПґуµДІъЖ·Ј¬ЅЁТй»®·ЦОЄІ»Н¬µДЧўІбµҐФЄЎЈЦьЛ®ПдЎўЖшМеОВ¶Иґ«ёРЖчЎўјУИИБ¬ЅУПЯµИІїјюїЙТФєНЦч»ъ»®·ЦОЄН¬Т»ёцЧўІбµҐФЄЎЈ
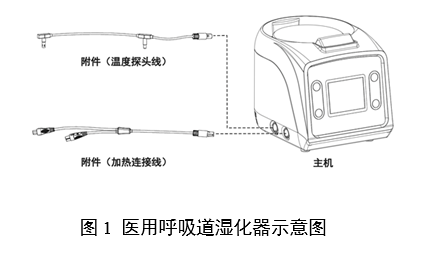
ЎЎЎЎ(¶ю)ТЅБЖЖчРµІъЖ·ЧўІбЧЫКцЧКБП
ЎЎЎЎ1.ІъЖ·ГиКц
ЎЎЎЎ1.1ГиКцІъЖ·µДЅб№№ЧйіЙЎЈТЅУГєфОьµАКЄ»ЇЖчІъЖ·Т»°гУЙЦч»ъЎўёЅјю(ИзУР)ЧйіЙЈ¬ёщѕЭІъЖ·КµјКЗйїцМṩІъЖ·Цч»ъЎўёЅјюµДКµОпНјЎўНјКѕєНБ¬ЅУКѕТвНјЈ¬ЅбєПКµОпНјЎўНјКѕєНБ¬ЅУКѕТвНјЈ¬¶ФІъЖ·µДЅб№№ЧйіЙЅшРРПкѕЎГиКцЎЈГиКцµДДЪИЭ°ьАЁЦч»ъєНёЅјюµДЅб№№ЎўіЯґзЎўІДБПЎўЦШБїµИЎЈ
ЎЎЎЎЦч»ъТ»°гУЙОВ¶ИїШЦЖДЈїйєНУГ»§ЅзГжЧйіЙЎЈ
ЎЎЎЎ1.2ЅбєПУГ»§ЅзГжЈ¬¶ФІъЖ·ѕЯ±ёµД№¦ДЬЎўРФДЬєНБЩґІК№УГБчіМЅшРРГиКцЎЈ
ЎЎЎЎ1.3МṩІъЖ·µДјУИИКЄ»ЇїШЦЖФАнНјЈ¬ФАнНјУ¦ДЬМеПЦёч№ШјьІїјюРЕПўЈ¬°ьАЁОВ¶ИїШЦЖДЈїй(є¬ёчЦЦґ«ёРЖч)ЎўКЄ»ЇКТєНјУИИБ¬ЅУПЯµИЎЈЗ°КцТ»Р©ІїјюїЙДЬєНЦч»ъІ»ФЪН¬Т»ёцЧўІбµҐФЄДЪЈ¬ЙкЗлИЛИФѕЙУ¦ёГЅбєПХвР©Іїјю¶ФІъЖ·µД№¤ЧчФАнЅшРРГиКцЎЈ
ЎЎЎЎЅбєПјУИИКЄ»ЇїШЦЖФАнНјЈ¬ПкПёЛµГчЖшМеОВ¶ИєНКЄ¶ИµДїШЦЖЎўјаІвєН±ЁѕЇ(ИзУР)ФАнЈ¬¶ФІ»Н¬ЖшМеБчБїЎўІ»Н¬БЩґІіЎѕ°(ОЮґґЎўУРґґНЁЖшµИ)µИМхјюПВµДІъЖ·№¤ЧчФАнЅшРРПкПёГиКцЎЈ
ЎЎЎЎ2.ККУГ·¶О§ЎўЅыјЙЦ¤
ЎЎЎЎ2.1ККУГ·¶О§
ЎЎЎЎІъЖ·УГУЪКЄ»ЇКдЛНёш»јХЯµДєфОьЖшМеЎЈ
ЎЎЎЎККУГИЛИєЈєКУІъЖ·ЙијЖЈ¬їЙТФУГУЪіЙИЛЎў¶щНЇЎўУ¤¶щµИЎЈ
ЎЎЎЎККУГ»·ѕіЈєГчИ·ІъЖ·µДК№УГіЎЛщЈ¬ИзТЅФєЎўјТНҐµИК№УГіЎЛщЎЈ
ЎЎЎЎИз№ыІъЖ·їЙТФФЪМШКв»·ѕіК№УГЈ¬ТІУ¦ЅшТ»ІЅЛµГчЈ¬Изј±ѕИЧЄФЛЎўёЯєЈ°ОМхјюПВК№УГµИЎЈ
ЎЎЎЎ2.2ЅыјЙЦ¤ЈєФЭОґ·ўПЦЎЈ
ЎЎЎЎ3.І»БјКВјюАъК·јЗВј
ЎЎЎЎТЅУГєфОьµАКЄ»ЇЖчПа№ШµДІ»БјКВјю±ЁёжЦчТЄУРКЄ»Ї№ЮЛ®О»µНОґ±ЁѕЇ»тОу±ЁѕЇЎўКЄ»ЇЖчУлјУИИРНєфОь№ЬВ·ґнОуБЄєПК№УГµјЦВєфОь№ЬВ·№эИИЎў№ЬВ·ИЫ»ЇЎў»јХЯЙХЙЛЎўЧЕ»рµИЎЈ
ЎЎЎЎТЅБЖЖчРµЧўІб°мАнЙкЗлИЛУ¦№ШЧўІўКХјЇН¬АаІъЖ·ТФј°Йк±ЁІъЖ·ЧўІбЦЬЖЪДЪµДІ»БјКВјюАъК·јЗВјЈ¬їЙНЁ№э·ўІјµДІ»БјКВјюЧКБПївЦРІйСЇПаУ¦І»БјКВјюКэѕЭЎЈїЙТФІъЖ·ГыіЖОЄ·ЅПтїЄХ№ТЅБЖЖчРµІ»БјКВјюІйСЇЈ¬НЁ№эФЪТССЎФсµДКэѕЭївµДјмЛчТіГжКдИлФ¤ЖЪТЄЅшРРІйСЇµДТЅБЖЖчРµІъЖ·ГыіЖЈ¬НЁ№эПЮЦЖјмЛчК±јдЅшРРПа№ШІъЖ·І»БјКВјюРЕПўКХјЇЎЈ
ЎЎЎЎ(Иэ)·ЗБЩґІЧКБП
ЎЎЎЎ1.·зПХ№ЬАнЧКБП
ЎЎЎЎІъЖ·µД·зПХ№ЬАн±ЁёжУ¦·ыєПGB/T 42062Ў¶ТЅБЖЖчРµ ·зПХ№ЬАн¶ФТЅБЖЖчРµµДУ¦УГЎ·µДУР№ШТЄЗуЈ¬ЙуІйТЄµг°ьАЁЈє
ЎЎЎЎ1.1ІъЖ·°ІИ«РФМШХчЕР¶ЁКЗ·сЧјИ·(ТАѕЭYY/T 1437);
ЎЎЎЎ1.2ОЈє¦·ЦОцКЗ·сИ«Гж(ТАѕЭGB/T 42062ёЅВјC);
ЎЎЎЎ1.3·зПХїЙЅУКХЧјФтЈ¬ЅµµН·зПХµДґлК©ј°ІЙИЎґлК©єу·зПХµДїЙЅУКХіМ¶ИЈ¬КЗ·сУРРВµД·зПХІъЙъЎЈ
ЎЎЎЎТЅБЖЖчРµІъЖ·ЧўІб°мАнёщѕЭGB/T 42062Ў¶ТЅБЖЖчРµ ·зПХ№ЬАн¶ФТЅБЖЖчРµµДУ¦УГЎ·Ј¬¶Ф“ТЅУГєфОьµАКЄ»ЇЖч”ТСЦЄ»тїЙФ¤јыµД·зПХЅшРРЕР¶ЁЈ¬ІъЖ·ФЪЅшРР·зПХ·ЦОцК±ЦБЙЩУ¦°ьАЁТФПВµДЦчТЄОЈє¦(ПкјыёЅјю)Ј¬ЖуТµ»№У¦ёщѕЭЧФЙнІъЖ·МШµгИ·¶ЁЖдЛыОЈє¦ЎЈХл¶ФІъЖ·µДёчПо·зПХЈ¬ЖуТµУ¦ІЙИЎУ¦¶ФґлК©Ј¬И·±Ј·зПХЅµµЅїЙЅУКЬµДіМ¶ИЎЈ
ЎЎЎЎ2.ІъЖ·јјКхТЄЗуУ¦°ьАЁµДЦчТЄРФДЬЦё±к
ЎЎЎЎІ»Н¬µДТЅУГєфОьµАКЄ»ЇЖчІъЖ·ЖдІОКэёщѕЭЙијЖТЄЗу»бУРЛщЗш±рЎЈ±ѕЦёµјФФтБРіцґЛАаІъЖ·їЙДЬЙжј°µДЦШТЄРФДЬІОКэЈ¬ЧўІбЙкЗлИЛїЙёщѕЭЧФЙнІъЖ·µДјјКхМШµгЦЖ¶ЁРФДЬЦё±кµДѕЯМеТЄЗуЎЈИфЖуТµРыіЖІъЖ·»№ѕЯУРЖдЛы№¤ЧчДЈКЅУ¦Н¬К±їјВЗЦЖ¶ЁПаУ¦µДРФДЬєН№¦ДЬТЄЗуЎЈ
ЎЎЎЎ2.1Цч»ъјјКхЦё±кТЄЗу
ЎЎЎЎТФПВЦё±кТАѕЭYY 9706.274±кЧјТЄЗуБРіцЈ¬Иф±кЧјёьРВЈ¬ЙкЗлИЛУ¦ТэУГµ±ПВПЦРР±кЧјЎЈЙкЗлИЛТЄЗуИфёЯУЪ±кЧјїЙ°ґЙкЗлИЛТЄЗуЦґРРЎЈ
ЎЎЎЎ2.1.1ЖшМеБчБї·¶О§ј°КЄ»ЇКдіцЈєУ¦ГчИ·І»Н¬МхјюПВµДЖшМеБчБї·¶О§ј°¶ФУ¦µДЖшМеКЄ»ЇКдіц;
ЎЎЎЎ2.1.2±»ІвЖшМеО¶ȼ໤װЦГЈєОВ¶ИПФКѕ·¶О§ЈєЦБЙЩОЄ25-45ЎжЈ¬ѕ«¶ИЈє≤±2Ўж;
ЎЎЎЎ2.1.3ФЛРРФлТф(ѕаЙи±ё1ГЧ)Јє≤50dB(A);
ЎЎЎЎ2.1.4ІЩЧчХЯїЙЙиЦГІОКэЈєУ¦ГчИ·ІЩЧчХЯїЙЙиЦГµДІОКэЈ¬Ф¤ЖЪУГУЪУРґґНЁЖшЦОБЖµДЦБЙЩУ¦°ьАЁ»јХЯ¶ЛЖшМеОВ¶ИЙиЦГ;
ЎЎЎЎ2.1.5ІОКэЛш¶Ё№¦ДЬ(ИзККУГ)Јє°ьАЁКдЛНЖшМеОВ¶ИЛш¶Ё;
ЎЎЎЎ2.1.6Ф¤ИИК±јдЈєУ¦ГчИ·ЧоґуФ¤ИИК±јд;
ЎЎЎЎ2.1.7±ЁѕЇЧ°ЦГ(ИзККУГ)ЈєИз»јХЯ¶Лі¬ОВ±ЁѕЇЎў»јХЯ¶ЛµНОВ±ЁѕЇЎў»јХЯ¶ЛµНКЄ¶И±ЁѕЇЎўЛ®БїµН±ЁѕЇ»тИ±Л®±ЁѕЇЎўЦч»ъјУИИДЈїйі¬ОВ±ЁѕЇЈ¬У¦·ыєПYY 9706.108µД№ж¶ЁЎЈ
ЎЎЎЎ2.2ЦьЛ®ПдРФДЬЦё±кТЄЗу(ИзУР)
ЎЎЎЎ2.2.1ЦьЛ®ИЭБї
ЎЎЎЎУ¦ГчИ·ЧоґуЛ®О»ПЯµДИЭБї;
ЎЎЎЎ2.2.2ЅУН·
ЎЎЎЎФІЧ¶ЅУН·У¦·ыєПYY/T 1040.1ЅУН·ТЄЗу;
ЎЎЎЎ2.2.3ЧоґуіРКЬС№Б¦
ЎЎЎЎУ¦ГчИ·ЧоґуіРКЬС№Б¦Ј¬іЦРш3minЈ¬У¦ОЮїЄБСПЦПу;
ЎЎЎЎ2.2.4ТєМеГЬ·вРФ
ЎЎЎЎјУЛ®ЦБЧоґуЛ®О»ПЯК±Ј¬јУИлЧоґуіРКЬЖшС№Ј¬У¦ОЮЙшВ©ПЦПу;
ЎЎЎЎ2.2.5ОЮѕъ»тОўЙъОпПЮ¶И
ЎЎЎЎИфіці§КЗОЄОЮѕъЧґМ¬Ј¬ФтУ¦ЦЖ¶ЁОЮѕъЦё±кЎЈК№УГЗ°ОЮРиЗеЅаЎўПы¶ѕµД·ЗОЮѕъК№УГІъЖ·Ј¬У¦¶ФЖдОўЙъОпПЮ¶ИЅшРРЖАјЫЈ¬Н¬К±їЙІОїјЎ¶ЦР»ЄИЛГс№ІєН№ъТ©µдЎ·ЦРОўЙъОпПЮ¶ИТ©µдјмІй·ЁЅшРРјмІв;
ЎЎЎЎ»·СхТТНйІРБфБїЈєИфѕ»·СхТТНйГрѕъЈ¬Фт»·СхТТНйІРБфБїЦё±кУ¦·ыєПGB/T 16886.7µДТЄЗу;
ЎЎЎЎОўЙъОпПЮ¶ИПЮЦµТЄЗуЈєК№УГЗ°ОЮРиЗеЅаЎўПы¶ѕµД·ЗОЮѕъК№УГІъЖ·Ј¬І»µГјміцґуі¦ѕъИєЎўЦВІЎРФ»ЇЕ§ѕъ;ПёѕъЧЬКэУ¦≤200cfu/gЎўХжѕъѕъВдЧЬКэ≤100cfu/gЎЈ
ЎЎЎЎ2.3Инјю№¦ДЬ
ЎЎЎЎТЅБЖЖчРµЧўІбґъАнЧўІбИЛУ¦·ыєПЎ¶ТЅБЖЖчРµИнјюЧўІбЙуІйЦёµјФФтЎ·ЦРµЪѕЕХВЧўІбЙк±ЁЧКБПІ№ідЛµГчЦР№ШУЪІъЖ·јјКхТЄЗуРФДЬЦё±кµДЅЁТйДЪИЭЎЈ
ЎЎЎЎ2.4°ІИ«ТЄЗу
ЎЎЎЎІъЖ·У¦·ыєПGB 9706.1ЎўYY 9706.274ЎўYY 9706.102ЎўYY 9706.108(ИзУР)±кЧјµДТЄЗуЎЈ
ЎЎЎЎ¶ФУЪФ¤ЖЪФЪјТНҐ»·ѕі»тЖдЛы·ЗЧЁТµТЅБЖ»·ѕіПВК№УГµДЙи±ёЈ¬У¦·ыєПYY 9706.111±кЧјµДТЄЗуЎЈ
ЎЎЎЎ3.Н¬Т»ЧўІбµҐФЄДЪјмСйµдРНРФІъЖ·И·¶ЁФФт
ЎЎЎЎ3.1µдРНІъЖ·У¦КЗН¬Т»ЧўІбµҐФЄДЪДЬ№»ґъ±н±ѕµҐФЄДЪЖдЛыІъЖ·°ІИ«РФєНУРР§РФµДІъЖ·ЎЈ
ЎЎЎЎ3.2ІъЖ·µДјУИИКЄ»ЇїШЦЖЎўКЄ»Ї№ЮЅб№№ЙијЖµИТтЛШѕц¶ЁБЛІъЖ·µДРФДЬЈ¬Из№ыЗ°КцТтЛШІо±рЅПґуЈ¬У¦·Ц±рЅшРРСйЦ¤ЎЈ
ЎЎЎЎ4.СРѕїЧКБП
ЎЎЎЎ4.1ІъЖ·РФДЬСРѕї
ЎЎЎЎТЅБЖЖчРµІъЖ·ЧўІбґъАнЅЁТйЙкЗлИЛЅбєПYY 9706.274±кЧјїЄХ№ТФПВПа№ШСРѕїЈ¬МбЅ»ПаУ¦СРѕїЧКБПЈ¬°ьАЁСРѕї·Ѕ°ёєНСРѕї±ЁёжЎЈСРѕї·Ѕ°ёУ¦°ьАЁСРѕїДїµДЎўїЙЅУКЬ±кЧјЎўКФСй№эіМµИЎЈ
ЎЎЎЎКЄ»ЇКдіцРФДЬСРѕїЈєМбЅ»КЄ»ЇКдіцРФДЬСРѕїЈ¬СРѕїІъЖ·ФЪХэіЈК№УГК±ЙщіЖµДБчБїЎўЙиЦГЎў»·ѕіОВ¶ИєНЅшЖшїЪОВ¶ИєНКЄ¶И·¶О§ДЪЈ¬ФЪ»јХЯБ¬ЅУїЪµДКЄ»ЇКдіцРФДЬЎЈ
ЎЎЎЎОВ¶ИїШЦЖРФДЬСРѕїЈєМбЅ»ОВ¶ИїШЦЖРФДЬСРѕїЈ¬СРѕїІъЖ·Йи¶ЁОВ¶ИµДЧјИ·РФЈ¬СРѕїіхКјОВ¶И(23Ўж±2Ўж)µЅЙи¶ЁОВ¶ИЛщРиµДК±јдЎЈ
ЎЎЎЎО¶ȼ໤РФДЬСРѕїЈєИз№ыѕЯУРЖшМеО¶ȼ໤№¦ДЬЈ¬МбЅ»О¶ȼ໤ѕ«¶ИСРѕїЧКБПЎЈ
ЎЎЎЎєфОь№ЬВ·јУИИРФДЬСРѕїЈєИз№ыїЙТФїШЦЖєфОь№ЬВ·јУИИЛїµД№¤ЧчЈ¬МбЅ»ПаУ¦µДСРѕїЧКБПЎЈ
ЎЎЎЎ±ЁѕЇРФДЬСРѕїЈєИз№ыѕЯ±ё±ЁѕЇ№¦ДЬЈ¬МбЅ»±ЁѕЇРФДЬСРѕїЧКБПЎЈ
ЎЎЎЎОЈПХµД·А»¤СРѕїЈє·АЦ№і¬ОВµДСРѕїЈ¬·А»рСРѕїµИЎЈ
ЎЎЎЎ4.2ЙъОпПаИЭРФЖАјЫСРѕї
ЎЎЎЎЖАјЫУл»јХЯЦ±ЅУ»тјдЅУЅУґҐІї·ЦµДЙъОпПаИЭРФЈ¬АэИзЖшМеОВ¶Иґ«ёРЖчЎўКЄ»Ї№ЮµИЎЈЙъОпПаИЭРФЖАјЫїЙТФёщѕЭYY/T 1778.1їЄХ№ЎЈЙъОпС§ЖАјЫ№эіМЦРУ¦µ±ЧўЦШФЛУГТСУРРЕПў(°ьАЁІДБПЎўОДПЧЧКБПЎўМеНвєНМеДЪКФСйКэѕЭЎўБЩґІѕСйЎў°ІИ«К№УГµДАъК·µИ)ЎЈ
ЎЎЎЎЙъОпПаИЭРФЖАјЫСРѕїЧКБПУ¦µ±°ьАЁЈєЙъОпПаИЭРФЖАјЫµДТАѕЭєН·Ѕ·ЁЈ¬ІъЖ·ЛщУГІДБПµДГиКцј°УлИЛМеЅУґҐµДРФЦКЎўІДБПУ¦УГАъК·Ј¬КµК©»т»нГвЙъОпС§КФСйµДАнУЙєНВЫЦ¤Ј¬¶ФУЪПЦУРКэѕЭЎў°ІИ«К№УГАъК·»тКФСйЅб№ыµДЖАјЫЎЈ
ЎЎЎЎ4.3Грѕъ/Пы¶ѕ№¤ТХСРѕї
ЎЎЎЎЦХ¶ЛУГ»§Грѕъ/Пы¶ѕЈє¶ФУЪКЧґОК№УГЗ°РиТЄУГ»§Грѕъ/Пы¶ѕЎў»тХЯїЙФЩУГЖчРµµДФЩґОК№УГЗ°РиТЄУГ»§Грѕъ/Пы¶ѕµДЈ¬У¦µ±ГчИ·НЖјцµДПы¶ѕјБЎўПы¶ѕ№¤ТХ(·Ѕ·ЁєНІОКэ)ТФј°ЛщНЖјцПы¶ѕ·Ѕ·ЁИ·¶ЁµДТАѕЭЈ¬ІўЗТУ¦МṩЛщНЖјцµДГрѕъ/Пы¶ѕ№¤ТХУРР§РФµД·ЦОцЖАјЫЎЈґЛНвЈ¬У¦Мṩ¶аґОЦШёґґ¦Ан¶ФЙи±ё°ІИ«ј°УРР§РФµДУ°ПмµДСРѕїЧКБПЎЈ
ЎЎЎЎІРБф¶ѕРФЈєИзГрѕъ/Пы¶ѕК№УГµД·Ѕ·ЁїЙДЬ»біцПЦІРБфЈ¬У¦ГчИ·ІРБфОпРЕПўј°ІЙИЎµДґ¦Ан·Ѕ·ЁІўМṩСРѕїЧКБПЎЈ
ЎЎЎЎ4.4ІъЖ·УРР§ЖЪєН°ьЧ°СРѕї
ЎЎЎЎІОїјЎ¶УРФґТЅБЖЖчРµК№УГЖЪПЮЧўІбјјКхЙуІйЦёµјФФтЎ·їЄХ№ІъЖ·К№УГЖЪПЮµДСРѕїЎЈ
ЎЎЎЎ»хјЬКЩГь(ИфККУГ)Јє¶ФУЪКЄ»ЇЖчµДёЅјюЈ¬Мṩ»хјЬКЩГьµДСРѕїЧКБПЎЈ¶ФУЪТ»ґОРФК№УГёЅјюЈ¬МṩЧоі¤К№УГК±јдµДСРѕїЧКБПЎЈ
ЎЎЎЎ¶ФУЪУРПЮґОЦШёґК№УГµДКЄ»ЇЖчµДёЅјюЈ¬МṩʹУГґОКэµДСРѕїЧКБПЎЈ
ЎЎЎЎХл¶ФРиТЄЗеПґПы¶ѕµДКЄ»ЇЖчµДІїјюЈ¬У¦їјВЗЗеПґПы¶ѕ¶ФК№УГЖЪПЮµДУ°ПмЎЈИзМṩʪ»Ї№Ю(ИзККУГ)ЎўЖшМеОВ¶ИјаІвґ«ёРЖч(ИзККУГ)УРПЮґОК№УГІїјюµДК№УГґОКэСйЦ¤ЧКБПЎЈїЙТФґУѕ«¶ИµДЅµµНКЗ·с»бУ°ПмµЅКЄ»ЇЖчµД»щ±ѕ°ІИ«Ул»щ±ѕРФДЬИҐ·ЦОцЧчОЄНЈУГµДМхјюЎЈ
ЎЎЎЎ°ьЧ°ј°°ьЧ°НкХыРФЈєФЪРыіЖµД»хјЬКЩГьЖЪДЪТФј°ФЛКдґўґжМхјюПВЈ¬±ЈіЦ°ьЧ°НкХыРФµДТАѕЭТФј°±ЈіЦІъЖ·РФДЬµД·ЦОцСРѕїЈ¬У¦МШ±рСРѕїФЪ»хјЬКЩГьЖЪЅбКшТФј°ѕ№эРыіЖДНКЬµДФЛКдґўґжМхјюПВ¶ФІъЖ·РФДЬµДУ°ПмЎЈ
ЎЎЎЎ4.5ИнјюСРѕї
ЎЎЎЎИнјю°ІИ«ј¶±рУ¦ЦБЙЩ№йОЄЦРµИЈ¬°ґХХЎ¶ТЅБЖЖчРµИнјюЧўІбЙуІйЦёµјФФтЎ·єНЎ¶ТЅБЖЖчРµНшВз°ІИ«ЧўІбЙуІйЦёµјФФтЎ·(ИзККУГ)µДПа№ШТЄЗуМбЅ»ІъЖ·ИнјюСРѕїЧКБПЎЈ
ЎЎЎЎ4.6ЖдЛыСРѕї(ИзККУГ)
ЎЎЎЎІъЖ·ѕЯУРРВјјКхМШРФК±Ј¬У¦МбЅ»Па№ШСРѕїЧКБПЈ¬ТФЦ¤ГчІъЖ·µД°ІИ«РФєНУРР§РФЎЈ
ЎЎЎЎ5.ЖдЛыЧКБП
ЎЎЎЎёГІъЖ·ОЄГвУЪЅшРРБЩґІЖАјЫµДТЅБЖЖчРµЎЈЙкЗлИЛ°ґХХЎ¶БРИлГвУЪБЩґІЖАјЫТЅБЖЖчРµДїВјІъЖ·¶Ф±ИЛµГчјјКхЦёµјФФтЎ·Ј¬ґУ»щ±ѕФАнЎўЅб№№ЧйіЙЎўРФДЬЎў°ІИ«РФЎўККУГ·¶О§µИ·ЅГжЈ¬Ц¤ГчІъЖ·µД°ІИ«УРР§РФЎЈ
ЎЎЎЎ(ЛД)БЩґІЖАјЫЧКБП
ЎЎЎЎёГІъЖ·БРИлЎ¶ГвУЪБЩґІЖАјЫТЅБЖЖчРµДїВјЎ·Ј¬ЙкЗлИЛОЮРиМбЅ»БЩґІЖАјЫЧКБПЎЈИфОЮ·ЁЦ¤ГчЙк±ЁІъЖ·УлЎ¶ГвУЪБЩґІЖАјЫТЅБЖЖчРµДїВјЎ·ЛщКцµДІъЖ·ѕЯУР»щ±ѕµИН¬РФЈ¬У¦°ґХХЎ¶ТЅБЖЖчРµБЩґІЖАјЫјјКхЦёµјФФтЎ·МбЅ»БЩґІЖАјЫЧКБПЎЈ
ЎЎЎЎ(Ое)ЛµГчКйєН±кЗ©Сщёе
ЎЎЎЎІъЖ·ЛµГчКйєН±кЗ©У¦µ±·ыєПЎ¶ТЅБЖЖчРµЛµГчКйєН±кЗ©№ЬАн№ж¶ЁЎ·єНGB 9706.1ЎўYY 9706.102ЎўYY 9706.274ТФј°ЖдЛыККУГ±кЧјЦР№ШУЪЛµГчКйєН±кЗ©µДПа№ШТЄЗуЎЈ
ЎЎЎЎН¬К±№ШЧўТФПВДЪИЭЈє
ЎЎЎЎ1.У¦ГчИ·ёГЙи±ёФЪѕ№эЕаСµµДТЅОсИЛФ±ЦёµјПВК№УГЎЈ
ЎЎЎЎ2.У¦ГчИ·Йи±ёµДК№УГ»·ѕіІўУ¦ёжЦЄІ»ФЪ№ж¶ЁµД»·ѕіМхјюПВК№УГїЙДЬµјЦВµД·зПХЎЈАэИзЈ¬»·ѕіОВ¶ИТЄЗуЎўµзФґТЄЗуµИ;ККУГµДєЈ°ОёЯ¶ИЎўКЗ·сККєПФЪё»Сх»·ѕіК№УГµИЎЈ
ЎЎЎЎ3.ИфІ»їЙФЪѕЯУРТЧИјЖшМеЎўё»Сх»·ѕіЦРК№УГЈ¬У¦ѕЇёжЙи±ёѕаАлТЧИјЖшМе»тСхФґЦБЙЩ1ГЧЎЈ
ЎЎЎЎ4.У¦ГчИ·Йи±ёІ»їЙФЪГ»УРЖшФґµДЗйїцПВК№УГЎЈ
ЎЎЎЎ5.У¦ГчИ·¶ФІЩЧчИЛФ±µДТЄЗуЎЈ
ЎЎЎЎ6.У¦ГчИ·К№УГК±Ј¬Йи±ёµД·ЕЦГёЯ¶ИТЄµНУЪ»јХЯєфОьµАЎЈ
ЎЎЎЎ7.У¦ГчИ·ёжЦЄІъЖ·ЛщККЕдµДєфОь№ЬВ·µД№жёс(ЅУїЪЦ±ѕ¶Ўў№ЬВ·і¤¶ИЎўТФј°¶ФУЪїЙБ¬ЅУјУИИєфОь№ЬВ·µДКЄ»ЇЖчёжЦЄУГ»§ККєПБ¬ЅУµДєфОь№ЬВ·µД¶о¶Ё№¦ВКј°КдіцµзС№)ЎЈ
ЎЎЎЎ8.У¦ГчИ·ёшіцЙи±ёТФј°ёЅјюµДЗеЅа»тПы¶ѕ·Ѕ·Ё(°ьАЁКЧґОК№УГєНЦШёґК№УГК±)ј°±ШТЄµДґ¦АнЎўЗеПґЦЬЖЪЎўёь»»ЦЬЖЪЎўёь»»ЛµГчЎўЧоґуїЙЦШёґЗеПґ/Пы¶ѕµДґОКэТФј°І»ДЬФЩК№УГµДЕР¶Ё±кЧјЎЈ
ЎЎЎЎ9.У¦ГчИ·ИнјюµД№¦ДЬЎўК№УГПЮЦЖЎўКдИлКдіцКэѕЭАаРНЎў±Ш±ёИнУІјюЎўЧоґуІў·ўКэЎўЅУїЪЎў·ГОКїШЦЖЎўФЛРР»·ѕі(ИфККУГ)ЎўРФДЬР§ВК(ИфККУГ)µИРЕПўЈ¬ГчИ·Инјю·ўІј°ж±ѕЎЈ
ЎЎЎЎ(Бщ)ЦКБї№ЬАнМеПµОДјю
ЎЎЎЎ1.ІъЖ·ЙъІъЦЖФмПа№ШТЄЗу
ЎЎЎЎУ¦µ±ГчИ·ІъЖ·ЙъІъ№¤ТХ№эіМЈ¬№¤ТХ№эіМїЙІЙУГБчіМНјµДРОКЅЈ¬ЗТУ¦ЅбєПІъЖ·КµјКЙъІъ№эіМПё»ЇІъЖ·ЙъІъ№¤ТХЅйЙЬЈ¬У¦ДЬМеПЦіцНвРјУ№¤Ії·Ц(ИзУР)Ўў°ліЙЖ·јУ№¤№эіМЎЈ№¤ТХБчіМНјЦРУ¦ГчКѕ№Шјь№¤РтЎўМШКв№эіМ(ИзУР)Ўў№эіМїШЦЖµгЎўёч№¤Рт¶Ф»·ѕіµДТЄЗуЎўК№УГµДПа№ШЙи±ёТФј°¶ФЙи±ёѕ«¶ИµДТЄЗуµИЈ¬ІўЛµГчЖдГїµА№¤РтµДІЩЧчЛµГчј°ЅУКХєН·ЕРР±кЧјЈ¬Н¬К±¶Ф№эіМїШЦЖТЄµгЅшРРПкПёЛµГчЎЈ
ЎЎЎЎ2.ЙъІъіЎµШ
ЎЎЎЎУ¦ПкПёЛµГчІъЖ·СРЦЖіЎµШЎўЙъІъіЎµШµШЦ·ЎўЙъІъ№¤ТХІјѕЦЎўЙъІъ»·ѕіТЄЗуј°ЦЬ±ЯЗйїцЎЈУР¶аёцСРЦЖЎўЙъІъіЎµШЈ¬У¦µ±ёЕКцГїёцСРЦЖЎўЙъІъіЎµШµДКµјКЗйїцЎЈ
ЙоЫЪєиФ¶ТЅБЖЖчРµЧЙСЇУРПЮ№«ЛѕКЗТ»јТјјКхЧЁТµµДТЅБЖЖчРµЧЙСЇ№«ЛѕЈ¬ЧЁЧўМṩȫ№ъёчµШИзЈєЙоЫЪЎў№гЦЭЎў¶«ЭёЎўЦРЙЅЎў·рЙЅЎўі±ЦЭЎўЛіµВЎў№гОчЎўЙПєЈЎўОч°ІЎўЦШЗмЎўіЙ¶јµИЦЄГыіЗКРµДТЅБЖЖчРµБмУтјјКхЧЙСЇ·юОсЎЈєиФ¶ТЅБЖЖчРµЧЙСЇЧЁТµ·юОсУЪЈєТЅБЖЖчРµІъЖ·ЧўІбЦ¤ЎўТЅБЖЖчРµЙъІъРнїЙЦ¤ЎўЅшїЪТЅБЖЖчРµЧўІбЎўТ»АаТЅБЖЖчРµІъЖ·±ё°ёј°ЙъІъ±ё°ёґъ°мЎўТЅБЖЖчРµѕУЄРнїЙЦ¤ґъ°мЎў¶юАаТЅБЖЖчРµѕУЄ±ё°ёЎўТЅБЖЖчРµ·ЦАаЅз¶ЁЎўCEИПЦ¤ЎўISO13485ИПЦ¤ЎўFDAЧўІбЎўБЩґІКФСйЎўТЅБЖЖчРµЦКБї№ЬАнМеПµИПЦ¤ј°МеПµЅЁБўУл№эіМИ·ИПОДјюЅЁБў(ISO9001, ISO13485, GMP, CEЈ¬QSR820Ј¬CMDCAS);ІъЖ·јјКхТЄЗуЦЖ¶©ЎўјјКхОДјюЎўБЩґІКФСйј°ГвБЩґІН¬АаІъЖ·±И¶Ф±ИЧКБП±аРґЎўЧўІбЧКБП±аРґёЁµјЎўµзґЕјжИЭФ¤ІвХыёДЎўТЅБЖЖчРµ№гёжЕъОДЙк±ЁЎўТЅБЖЖчРµіцїЪПъКЫЦ¤Гч°мАнЎўЅаѕ»КТЅЁЙиЦёµјµИМṩһվʽ·юОсЈ¬јјКхЧЁТµЈ¬іПРЕ·юОсЈ¬»¶УДъЧЙСЇЎЈ
ЎЎЎЎ
|

