|
ќ£Ї¶µƒЈ÷ја |
ќ£Ї¶µƒ–ќ≥…‘≠“т |
њ…ƒ№µƒЇуєы |
|
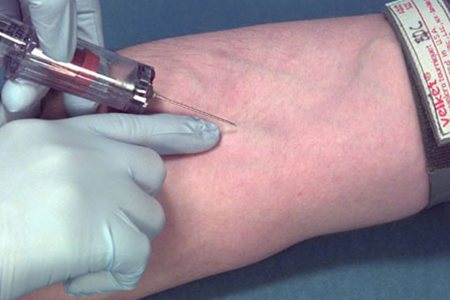
…ъќп—Іќ£Ї¶ |
≤ъ∆Јќіљш––√рЊъїт√рЊъ≤ї≥єµ„ |
‘м≥…≤ъ∆Јќџ»Њ£ђ‘Џ є”√єэ≥ћ÷–»зЈҐ…ъ—™“ЇƒжЅчµЉ÷¬≤°»ЋЈҐ…ъ∞№—™÷Ґ°£ |
≤ъ∆Ј≤їЊ≠√рЊъїтќііпµљ‘§∆Џµƒ√рЊъ–Ієы
є§“’ЇЌ…ъ≤ъїЈЊ≥≤їµ±
Ћщ”√µƒ»№љвЋЃіњґ»√їіпµљ“™«у |
≤…—™є№ƒЏ“ЇћеЈҐ…ъ√є±д |
|
≤ъ∆Јќі‘Џ«ељаїЈЊ≥÷–…ъ≤ъ |
“э»лѕЄЊъƒЏґЊЋЎїтќҐЅ£ќп÷ |
|
‘≠≤ƒЅѕ÷ Ѕњ≤їЇѕЄс |
≤ъ∆ЈƒЏ±ЏЈҐ…ъЋЃљвґш≤ъ…ъЌ—¬д |
|
”л≤ъ∆Ј є”√ѕаєЎµƒќ£Ї¶ |
ћнЉ”ЉЅЈ÷≤Љ≤їЊщ‘» |
ґ‘—™—щ≤ъ…ъ”∞ѕмґшµЉ÷¬Љм—йљбєы≤ї„Љ»Ј |
|
≤ї’э≥£ є”√їт≤…—™є№ЈҐ…ъ∆∆Ѕ— |
—™“ЇЈҐ…ъƒжЅчЄшїЉ’я‘м≥……ЋЇ¶
Єш≤ў„ч’яішјі“вЌв…ЋЇ¶ |
|
їЈЊ≥—єЅ¶±дїѓ |
µЉ÷¬≤…—™Ѕњ≤ї„Љ»Јїт≤ї„г |
|
≤…—™∆ч≤ƒ£®≤…—™’л£©ѕыЇƒєэґа’жњ’ґ» |
µЉ÷¬≤…—™Ѕњ≤ї„г |
|
‘§≥й’жњ’≤ї„Љ»Јїт–є¬© |
µЉ÷¬≤…—™Ѕњ≤ї„Љ»Ј |